Abstract
-
Background
- Overactivation of inflammatory cells, including macrophages and neutrophils, is associated with acute lung injury. BMS-470539 is a selective agonist of melanocortin 1 receptor, which triggers the inhibition of proinflammatory responses, suppressing neutrophil infiltration and protecting tissue. This study evaluated the effects of BMS-470539 on lipopolysaccharide-induced acute lung injury in a mouse model.
-
Methods
- Mice received a subcutaneous injection of saline or BMS-470539 (18.47 mg/kg) 1 hour before an intratracheal injection of saline or lipopolysaccharide (20 μg). Mice were sacrificed to analyze the severity of pulmonary edema (lung wet-to-dry weight [W/D] ratio) and inflammatory responses (level of leukocytes, polymorphonuclear neutrophils [PMNs] and tumor necrosis factor alpha [TNF-α] in bronchoalveolar lavage fluid [BALF]), and neutrophil infiltration (myeloperoxidase activity). TNF-α activation was also measured in neutrophils from bone marrow. Survival was investigated in a second-hit sepsis mouse model.
-
Results
- BMS-470539 improved sepsis-induced pulmonary edema, as demonstrated by a decreased W/D ratio (5.76%±0.83% to 3.81%±0.86%, P<0.05). The inflammatory response also improved, as shown by decreased levels of leukocytes (551±116 to 357±86×10²/mm³, P<0.05), PMNs (51.52%±16.23% to 18.41%±7.25%, P<0.01), and TNF-α (550±338 to 128±52 pg/ml, P<0.01) in the BALF. BMS-470539 also improved the inflammatory response, as shown by TNF-α levels (850±158 to 423±59 pg/ml, P<0.01) in neutrophils. BMS-470539 downregulated neutrophil infiltration in the lung (myeloperoxidase: 654±98 to 218±89 U/g, P<0.001). Lastly, BMS improved the survival rate (0% to 70%, P<0.01) in a mice multiple organ failure model.
-
Conclusions
- BMS-470539 improved lipopolysaccharide-induced acute lung injury and mortality in mice by affecting the inflammatory response.
-
Keywords: acute lung injury; cytokines; lipopolysaccharides; melanocortin 1 receptor
INTRODUCTION
Acute lung injury and acute respiratory distress syndrome (ALI/ARDS) is a complex syndrome consisting of an intense pulmonary inflammatory response with high morbidity and mortality. No specific treatment for ALI is available. Instead, patients generally receive supportive care, with options including mechanical ventilation, infection control, corticosteroid administration, and hemodynamic drugs; however, the survival benefits of those therapies are limited [1,2]. In early ALI, excessive inflammatory cell infiltration and inflammation occur, which contribute to pathological damage of the alveolar epithelium and vascular endothelium, an increase in capillary permeability, protein-rich cellular exudation, alveolar hemorrhage, and pulmonary hyaline membrane formation [1,3,4]. During this phase, inflammatory mediators such as cytokines (e.g., tumor necrosis factor alpha [TNF-α], interleukin 1, interleukin 8) are increased, and these pro-inflammatory cytokines play a crucial role in the progression of lung injury [4,5]. Thus, inhibiting excessive inflammatory cell transmigration or inflammation may be an effective treatment option for ALI.
The compound BMS-470539 binds to melanocortin 1 receptor (MC1R). The term “melanocortin” refers to a group of peptide hormones that includes α-, β-, and γ-melanocytestimulating hormone (MSH) and adrenocorticotropic hormone, which exert their effects by binding to and activating melanocortin receptors. Each receptor contributes to the diverse physiological functions of melanocortins, which involve pigmentation, steroidogenesis, energy homeostasis, exocrine secretion, sexual function, analgesia, inflammation, immunomodulation, temperature control, cardiovascular regulation, and neuromuscular regeneration [6-8]. MC1R activation has been implicated in the restoration of inflammatory response homeostasis, as it has been detected in various immune cells, including monocytes, neutrophils, macrophages, cytotoxic T cells, and dendritic cells [7,9]. In several studies, MC1R targeting has shown anti-inflammatory and immunomodulatory effects [7,10-14]. Among these studies, BMS-470539, a selective MC1R agonist, has emerged as a promising anti-inflammatory candidate that inhibits neutrophil recruitment and, leukocyte migration in the lung [7,10,12]. Therefore, in this study, we evaluated the effects of BMS-470539 on lipopolysaccharide-induced acute lung injury in a mouse model.
MATERIALS AND METHODS
Materials
BMS-470539 and Escherichia coli 055:B5 lipopolysaccharide were purchased from Sigma-Aldrich (St. Louis, MO, USA). Sevoflurane was obtained from Abbott Laboratories (Chicago, IL, USA). Immunoreactive TNF-α was quantified using commercially available enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Minneapolis, MN, USA), in accordance with the manufacturer’s instructions.
In Vitro Experiments
Cell culture and treatment
Mouse neutrophils were purified from bone marrow cell suspensions as described previously [12]. Briefly, to obtain a bone marrow cell suspension, a mouse femur and tibia were flushed with 5 ml of Roswell Park Memorial Institute 1,640/penicillin/streptomycin. The cells were passed through a glass wool column and were pelleted by centrifugation at 1,000 rpm for 10 minutes. The cell pellets were resuspended in 0.3% fetal calf serum/phosphate-buffered saline (PBS), and then neutrophils were purified from bone marrow cells using Histopaquebased density gradient centrifugation. Using Wright’s stained cytospin preparations, neutrophil purity was confirmed to be greater than 97%. Cell viability was determined by trypan blue exclusion and was consistently greater than 98%. To evaluate the effects of BMS-470539 on cytokine expression (TNF-α) in lipopolysaccharide-stimulated neutrophils, neutrophils were cultured with lipopolysaccharide (100 ng/ml) and BMS-470539 (at concentrations of 0, 1, 10, and 100 μM) dissolved in PBS for 4 hours.
Measurement of TNF-α
TNF-α levels in the neutrophil culture medium were quantified by ELISA kits obtained from R&D Systems according to the manufacturer’s instructions.
In Vivo Experiments
Animal model of lipopolysaccharide-induced acute lung injury
Male albino Institute of Cancer Research mice (30–35 g, 8 weeks old) were purchased from Samtako Science (Daejeon, Korea). The animals were kept on a 12-hour light/dark cycle with free access to water and food. All experiments were conducted according to Institutional Review Board and approved protocols (CNU IACUC-H-2016-27).
In the mice, ALI was induced by intratracheal administration of E. coli 055:B5 lipopolysaccharide. In this model, ALI was characterized by neutrophil infiltration into the lung interstitium and increased proinflammatory cytokine production after the lipopolysaccharide injection, with significant accumulation of neutrophils in the airways and histologic injury present at 24 hours after lipopolysaccharide exposure [15,16]. Mice were randomly assigned to one of four groups: mice receiving subcutaneous saline (0.1 ml) and intratracheal saline (40 μl) (control model [CON] group, n=10); mice receiving subcutaneous saline and intratracheal lipopolysaccharide (20 μg; 40 μl) (lipopolysaccharide group, n=10); mice receiving subcutaneous BMS-470539 (18.47 mg/kg; 0.1 ml) and intratracheal saline (BMS group, n=10); and mice receiving subcutaneous BMS-470539 (18.47 mg/kg; 0.1 ml) and intratracheal lipopolysaccharide (BMS+lipopolysaccharide group, n=10). The mice were initially anesthetized with sevoflurane. A subcutaneous injection of saline or BMS-470539 was performed 1 hour prior to the intratracheal injection of saline or lipopolysaccharide. In all 10 mice in each group, the lung wetto-dry weight (W/D) ratio was assessed, and myeloperoxidase and bronchoalveolar lavage fluid (BALF) were analyzed in five mice each. Animals were euthanized to collect BALF and lung tissues 18 hours after the intratracheal administration of saline or lipopolysaccharide (Figure 1). The selective agonist BMS-470539 (molecular weight=559.697 g/mol) was given at a dose, timing, and route of administration shown to be inhibitory in three distinct models of inflammation [7]. Therefore, a dose of 18.47 mg/kg (33 mmol/kg) was given subcutaneously in a fresh solution of PBS before inducing ALI.
Preparation of BALF
PBS (1 ml) was infused slowly into the trachea and withdrawn three times. Ethylenediaminetetraacetic acid–2Na was added to the BALF and cooled to 4°C to prevent leukocyte metabolism. The BALF was analyzed to assess cell differentiation. A cytocentrifuged spin preparation (CF-RD, Sakura, Tokyo, Japan) of the BALF was stained with the Wright–Giemsa stain to detect cell differentiation. The BALF was then centrifuged at 3,500 rpm at 4°C for 20 minutes to remove the cells. The cellfree supernatant was stored at −80°C for TNF-α measurements.
Lung W/D ratio
The left lung was weighed and then dried to a constant weight at 60°C over 7 days in an oven. To assess tissue edema, the W/D ratio was calculated.
Myeloperoxidase activity assay
Red blood cells were flushed from the lung vascular bed with an injection of 5 ml of ice-cold PBS through the right ventricle. The right lungs were removed, rinsed two times in ice-cold PBS, blotted dry, and snap-frozen in liquid nitrogen. The frozen lungs were then weighed and stored at 80°C if not used immediately. Frozen tissue was homogenized in 20 mM potassium phosphate buffer (pH 7.4) and then centrifuged for 30 minutes at 20,000 g. The pellet was resuspended in 50 mM potassium phosphate buffer (pH 6.0) containing 0.5% hexadecyltrimethylammonium bromide. Samples were sonicated and stored at 60°C for 2 hours. After incubation, 1 ml of each sample was centrifuged briefly, and the supernatant was assayed for myeloperoxidase activity in a hydrogen peroxide-Odianisidine buffer via spectrophotometric analysis at 420 nm. Results are given as units of myeloperoxidase activity per gram of lung tissue.
Survival and a multiple organ failure animal model
Mice were intraperitoneally administered E. coli 055:B5 lipopolysaccharide (5 mg/kg body weight) in order to induce the initial septic insult. A 24-hour post-lipopolysaccharide medium-grade cecal ligation and puncture (CLP) (25-gauge needle, double puncture) procedure was used to create a multiple organ failure (MOF) model. With the exception of a different method for lipopolysaccharide injection in the model, the surgical protocol was the same as previously published [17,18]. CLP was used to induce sepsis as described previously. First, the mice were anesthetized by isoflurane inhalation, and anesthesia was maintained throughout surgery. Next, the distal third of the cecum was ligated with a 3-0 sterile silk and punctured twice with a 25-gauge needle. Then, the cecum was gently squeezed to extrude a few pieces of feces before placing the cecum back into the abdomen. The abdominal wall and skin were closed in two layers, and 1 ml of pre-warmed lactated Ringer’s solution plus 5% dextrose with 0.05 mg/kg of buprenorphine was administered intraperitoneally after surgery. Mice experience a subacute inflammatory response after CLP with a 25-gauge needle in combination with lipopolysaccharide, which may lead to early MOF. To determine the effect of BMS-470539, all mice post-CLP were divided into two groups: (1) the MOF group and (2) the BMS group. At 4 hours post-CLP, the mice were injected intraperitoneally with 18.47 mg/kg body weight of BMS-470539. In the sham control model (CON), the mice underwent surgery, but their cecum was neither ligated nor punctured. All models were monitored twice daily for 14 days or until death.
Statistical Analysis
The data are presented as the mean±standard error of the mean for each experimental group. One-way analysis of variance and the Tukey-Kramer multiple-comparisons test (for multiple groups) or the Student t-test (for comparisons between two groups) was used. P-values less than 0.05 were considered to indicate statistical significance.
RESULTS
The Effects of BMS-470539 on Cytokine Expression (TNF-α) in Lipopolysaccharide-Stimulated Neutrophils
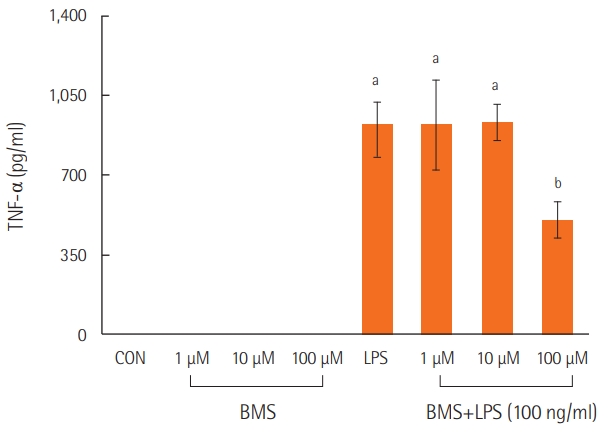
Neutrophils stimulated with lipopolysaccharide showed higher levels of TNF-α than those in the control or BMS groups. However, treatment with a combination of lipopolysaccharide and BMS-470539 significantly inhibited TNF-α expression in neutrophils, specifically at a BMS-470539 concentration of 100 μM (Figure 2).
Analysis of BALF
The concentrations of neutrophils and TNF-α in the BALF were significantly higher in the lipopolysaccharide group than in the control group, and BMS-470539 significantly attenuated the lipopolysaccharide-induced neutrophil infiltration and TNF-α production in the BALF (Table 1).
Myeloperoxidase Activity
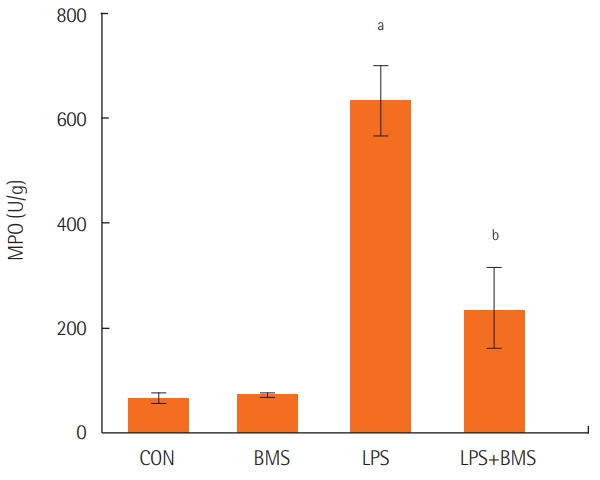
Myeloperoxidase levels were assayed in the lungs from control mice, from mice given BMS-470539 or lipopolysaccharide, and from mice pretreated with BMS-470539 before lipopolysaccharide administration. Lipopolysaccharide-induced neutrophil infiltration in the lung was lower in the group treated with BMS-470539 (Figure 3).
Lung W/D Ratio
The lung W/D ratio was higher in the lipopolysaccharide group than in the control group. A smaller increase in the W/D ratio was found in the group treated with BMS-470539 (Figure 4).
Mortality in a MOF Animal Model
Mortality in the MOF group was 80% on day 4 and 100% on day 8. By contrast, mortality in the BMS-470539-treated group was 0% on day 4 and 30% on day 8 (Figure 5).
DISCUSSION
The purpose of this study was to explore the effects of BMS-470539 on lipopolysaccharide-induced ALI. Our principal finding was that BMS attenuated the lipopolysaccharide-induced ALI, as determined by the W/D ratio and mortality in the MOF model. These findings seemed to be associated with the anti-inflammatory properties of BMS, including attenuation of TNF-α production via inhibition of neutrophil activation in response to lipopolysaccharide stimulation in both in vitro and in vivo conditions.
Lipopolysaccharide-induced ALI involves a highly complex inflammatory cascade characterized by the release of inflammatory cytokines and influx of inflammatory cells into the lung tissue [19]. TNF-α is one of the most important cellular pro-inflammatory factors that causes ALI. As an early proinflammatory cytokine, TNF-α is known to trigger the activation of other proinflammatory cytokines, such as interleukin-6 and interleukin-8, which enhance the inflammatory reaction. These can directly damage endothelial cells and increase their permeability. Furthermore, TNF-α can activate the release of protease and oxidation substances and inhibit surfactant activity on the alveolar surface, promoting a reduction of pulmonary compliance and pulmonary edema [20]. In ALI, neutrophils are the first immune cells recruited to the site of injury. In the early stage of lung injury, changes occur in the deformability of neutrophils, and they become entrapped in the pulmonary capillaries. Neutrophil entrapment is followed by permeability changes and edema formation [4]. Therefore, TNF-α and neutrophils play an important role in the mechanism of ALI.
As mentioned previously, BMS-470539 is a highly selective small-molecule agonist of MC1R. MC1R is the “classical” melanocyte α-MSH receptor that is expressed in melanocytes, monocytes, mast cells, and neutrophils, and is involved in blocking inflammation and augmenting pigmentation [6,8]. The mechanisms underlying the anti-inflammatory effects of α-MSH are highly diverse and complex. According to previous reports, α-MSH exerts its anti-inflammatory effects by inhibiting nitric oxide synthesis in murine macrophages [21], by inhibiting neutrophil migration to inflammatory sites in vitro in human neutrophils [22], by reducing the release of TNF-α and interleukin-1 in lipopolysaccharide-induced mouse serum [23], and by upregulating interleukin-10 in human monocytes [24]. Several reports have suggested that peptide compounds specific for MC1R exert anti-inflammatory effects that are reduced by α-MSH [7,10,11,13,14]. Thus, we evaluated the antiinflammatory effects of BMS-470539, a selective MC1R agonist peptide, on lipopolysaccharide-induced ALI.
In this experiment, the effect of BMS-470539 on the degree of ALI caused by an endotoxin was assessed in actual animal models through measurements of the W/D ratio. These findings may provide further evidence of the anti-inflammatory properties of BMS-470539, which include the attenuation of TNF-α production by inhibiting the activation of neutrophils stimulated with lipopolysaccharide in both in vitro and in vivo conditions. These findings suggest that BMS-470539 significantly attenuated lipopolysaccharide-induced neutrophil activation and TNF-α production in the BALF, and reduced lipopolysaccharide-induced neutrophil infiltration in the lung. Additionally, to investigate the association of neutrophil activation with inflammatory cytokines, normal neutrophils were isolated from the bone marrow of mice. A previous study showed that BMS-470539 reduced lipopolysaccharide-induced TNF-α accumulation in a dose-dependent manner in an in vivo study, with an 50% effective dose between 3.7 and 11.1 μmol/kg [7]. Unlike that in vivo study, we performed an in vitro study, gradually increasing the dose from 0 μM of BMS-470539, and showed an inhibitory effect on lipopolysaccharide-induced TNF-α production at 100 μM. Based on the previous experimental results, we concluded that the effect of BMS-470539 pretreatment in ALI was related to the regulation of neutrophil responses to the endotoxin. Because extrapulmonary ARDS due to sepsis also plays an important role in neutrophil function [1], we experimented with BMS-470539 to investigate its possible therapeutic effect in a CLP-induced MOF model, reflecting the conditions associated with the highest mortality in clinical practice. In that experiment, mortality was lower in the BMS-470539–treated group than in the control group.
The main limitation of our study is that we did not investigate the effects of BMS-470539 on other immune cells because of our focus on its effects on neutrophil and inflammatory cytokines (TNF-α) in ALI. In previous studies, the effects of α-MSH stimulating agents on immune cells have been reported [21-24], and to clarify the anti-inflammatory effect of BMS-470539, further studies exploring the effects of BMS-470539 on other immune cells are needed. Another limitation is that we did not perform an autopsy to confirm the presence of ALI in the CLP-induced sepsis model. Nonetheless, in severe sepsis, death is caused by MOF, and the respiratory system is known to be the most intensely affected organ system [1]. Our results showed that the mortality rate was reduced by BMS-470539 in the CLP-induced MOF model, but further experiments should be performed to determine whether this outcome was caused by the effects of BMS-470539 on ALI.
In conclusion, this study showed that BMS-470539, a selective small-molecule agonist of MC1R, suppressed TNF-α production by inhibiting neutrophil activation in a murine model of lipopolysaccharide-induced ALI. In our opinion, these results suggest that BMS-470539 may be effective in attenuating ALI due to sepsis, but further study is needed to establish its effects conclusively.
KEY MESSAGES
▪BMS-470539 is a selective agonist of melanocortin 1 receptor.
▪This study showed that BMS-470539 suppressed tumor necrosis factor alpha production by inhibiting neutrophil activation in a lipopolysaccharide-induced acute lung injury murine model.
▪BMS-470539 improved lipopolysaccharide-induced acute lung injury and mortality in mice by affecting the inflammatory response.
NOTES
-
CONFLICT OF INTEREST
No potential conflict of interest relevant to this article was reported.
-
AUTHOR CONTRIBUTIONS
Conceptualization: SHK. Data curation: JYK, TDT. Formal analysis: EAJ, JYK, TDT. Funding acquisition: SHK. Methodology: SHL, SHK. Project administration: SHK. Visualization: JAS, EAJ. Writing – original draft: EAJ, TDT. Writing – review & editing: EAJ, SHL, SHK.
Acknowledgments
This study was supported by a grant (CRI14001-1) from the Chonnam National University Hospital Research Institute of Clinical Medicine.
Figure 1.(A, B) Animal model of LPS-induced acute lung injury. Mice were randomly assigned to one of four groups: mice receiving subcutaneous (SC) saline (0.1 ml) and intratracheal (IT) saline (40 μl) (control model [CON] group, n=10); mice receiving SC saline and IT lipopolysaccharide (LPS) 20 μg (40 μl) (LPS group, n=10); mice receiving SC BMS-470539 18.47 mg/kg (0.1 ml) and IT saline (BMS group, n=10); mice receiving SC BMS-470539 18.47 mg/kg (0.1 ml) and IT LPS (BMS+LPS group, n=10). Mice were initially anesthetized with sevoflurane. The SC injection of saline or BMS performed 1 hour prior to the IT injection of saline or LPS. Animals were euthanized to collect the BALF and lung tissues at 18 hours after the IT administration of saline or LPS. PBS: phosphate-buffered saline; BALF: bronchoalveolar lavage fluid.

Figure 2.The effects of BMS-470539 on cytokine expression (TNF-α) in LPS-stimulated neutrophils. Neutrophils from mouse bone marrow were incubated with the indicated concentration of BMS470539 with or without LPS (100 ng/ml) for 4 hours, and then TNF-α levels were measured in the culture supernatants by ELISA. Each bar represents the mean±standard error of the mean. Data are representative of three separate experiments. TNF-α: tumor
necrosis factor alpha; CON: control model; LPS: lipopolysaccharide; ELISA: enzyme-linked immunosorbent assay. aP<0.01, vs. CON; bP<0.01, vs. LPS.
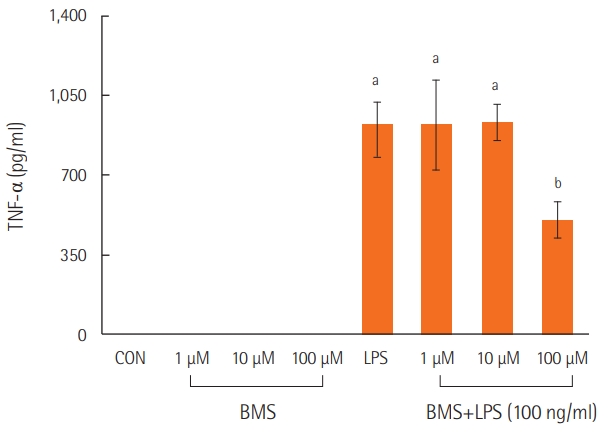
Figure 3.The effects of BMS-470539 on MPO content in the right lung. MPO was measured in the right lungs of the control mice (CON) and mice treated with BMS-470539 (18.47 mg/kg) alone (BMS), lipopolysaccharide (20 μg) alone (LPS), or BMS-470539 (18.47 mg/kg) before lipopolysaccharide (20 μg) (BMS+LPS). The data are from five mice in each group. Each bar represents the mean±standard error of the mean. MPO: myeloperoxidase. aP<0.001, vs. CON; bP<0.001 vs. LPS.
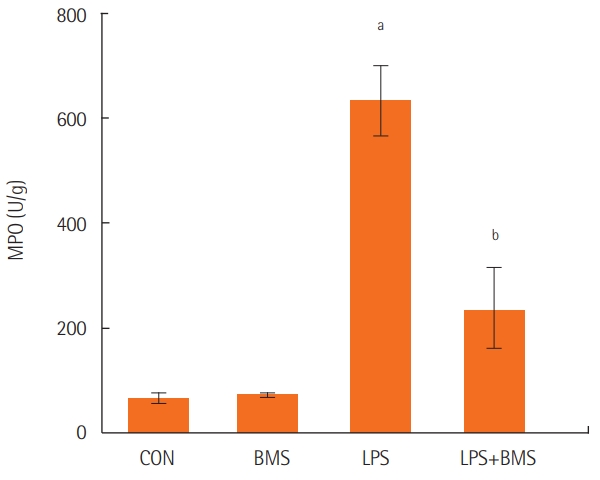
Figure 4.The effects of BMS-470539 on the wet-to-dry weight (W/D) ratio in the left lung. The W/D ratio was measured in the left lungs of the control mice (CON) and mice treated with BMS-470539 (18.47 mg/kg) alone (BMS), lipopolysaccharide (20 μg) alone (LPS), or BMS-470539 (18.47 mg/kg) before lipopolysaccharide (20 μg) (BMS+LPS). The data are from five mice in each group. Each bar represents the mean±standard error of the mean. aP<0.01, vs. CON; bP<0.05, vs. LPS.
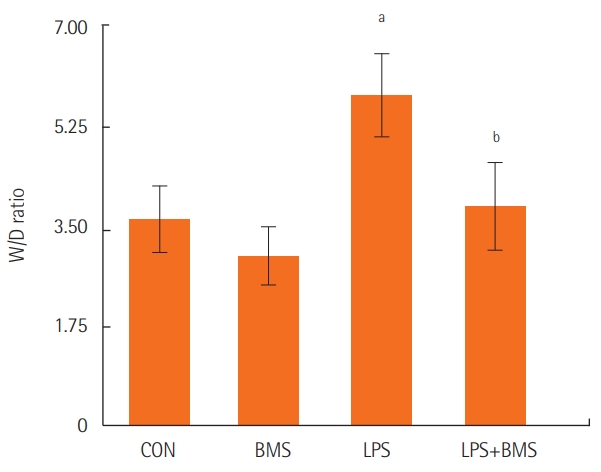
Figure 5.The effects of BMS-470539 on mortality in a multiple organ failure (MOF) animal model. Survival (10 mice per group) was monitored for 14 days after establishment of the MOF model. Mice were intraperitoneally administered Escherichia coli 055:B5 lipopolysaccharide (LPS; 5 mg/kg body weight) to induce the initial septic insult, and at 24 hours post-LPS administration, a medium-grade cecal ligation and puncture (25-gauge needle, double puncture) procedure was performed to create the MOF model. To determine the effects of BMS-470539, all mice that received cecal ligation and puncture were divided into two groups: (1) the MOF group, and (2) the BMS group: at 24 hours post-LPS administration, the mice received an intraperitoneal injection of BMS-470539 (18.47 mg/kg body weight). In the sham control model, the mice were opened abdominally to perform the same surgical procedure, but their cecum was neither ligated nor punctured. All models were monitored twice daily for 14 days or until mortality.
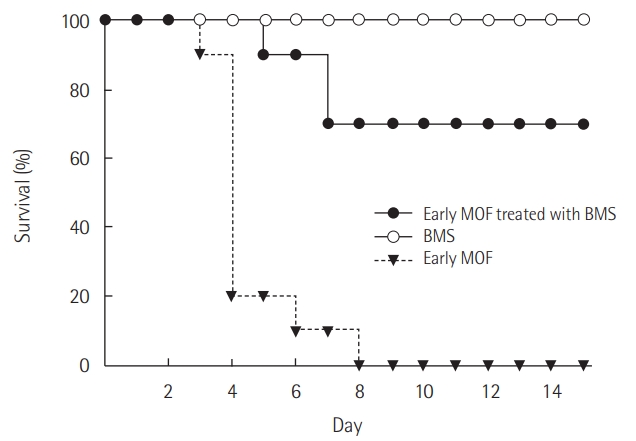
Table 1.Immune and inflammatory responses in BALF
|
Variable |
CON |
LPS 20 μg |
BMS 18.47 mg/kg |
BMS 18.47 mg/kg+LPS 20 μg |
|
Leukocyte (×102/mm3) |
251±46 |
551±116a
|
231±52 |
357±86b
|
|
PMN/total cell (%) |
3.52±0.23 |
51.52±16.23a
|
4.12±0.33 |
18.41±7.25b
|
|
TNF-α (pg/ml) |
15±34 |
550±338a
|
2±3 |
128±52b
|
References
- 1. Wheeler AP, Bernard GR. Acute lung injury and the acute respiratory distress syndrome: a clinical review. Lancet 2007;369:1553-64.ArticlePubMed
- 2. Bellani G, Laffey JG, Pham T, Fan E, Brochard L, Esteban A, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA 2016;315:788-800.ArticlePubMed
- 3. Dushianthan A, Grocott MP, Postle AD, Cusack R. Acute respiratory distress syndrome and acute lung injury. Postgrad Med J 2011;87:612-22.ArticlePubMed
- 4. Grommes J, Soehnlein O. Contribution of neutrophils to acute lung injury. Mol Med 2011;17:293-307.ArticlePubMedPDF
- 5. Chopra M, Reuben JS, Sharma AC. Acute lung injury: apoptosis and signaling mechanisms. Exp Biol Med (Maywood) 2009;234:361-71.ArticlePubMed
- 6. Wikberg JE, Muceniece R, Mandrika I, Prusis P, Lindblom J, Post C, et al. New aspects on the melanocortins and their receptors. Pharmacol Res 2000;42:393-420.ArticlePubMed
- 7. Kang L, McIntyre KW, Gillooly KM, Yang Y, Haycock J, Roberts S, et al. A selective small molecule agonist of the melanocortin-1 receptor inhibits lipopolysaccharide-induced cytokine accumulation and leukocyte infiltration in mice. J Leukoc Biol 2006;80:897-904.ArticlePubMed
- 8. Gantz I, Fong TM. The melanocortin system. Am J Physiol Endocrinol Metab 2003;284:E468-74.ArticlePubMed
- 9. Maaser C, Kannengiesser K, Specht C, Lügering A, Brzoska T, Luger TA, et al. Crucial role of the melanocortin receptor MC1R in experimental colitis. Gut 2006;55:1415-22.ArticlePubMedPMC
- 10. Leoni G, Voisin MB, Carlson K, Getting S, Nourshargh S, Perretti M. The melanocortin MC(1) receptor agonist BMS-470539 inhibits leucocyte trafficking in the inflamed vasculature. Br J Pharmacol 2010;160:171-80.ArticlePubMedPMC
- 11. Chen W, Li J, Qu H, Song Z, Yang Z, Huo J, et al. The melanocortin 1 receptor (MC1R) inhibits the inflammatory response in Raw 264.7 cells and atopic dermatitis (AD) mouse model. Mol Biol Rep 2013;40:1987-96.ArticlePubMedPDF
- 12. Holloway PM, Durrenberger PF, Trutschl M, Cvek U, Cooper D, Orr AW, et al. Both MC1 and MC3 receptors provide protection from cerebral ischemia-reperfusion-induced neutrophil recruitment. Arterioscler Thromb Vasc Biol 2015;35:1936-44.ArticlePubMedPMC
- 13. Mykicki N, Herrmann AM, Schwab N, Deenen R, Sparwasser T, Limmer A, et al. Melanocortin-1 receptor activation is neuroprotective in mouse models of neuroinflammatory disease. Sci Transl Med 2016;8:362ra146. ArticlePubMed
- 14. Qiang X, Liotta AS, Shiloach J, Gutierrez JC, Wang H, Ochani M, et al. New melanocortin-like peptide of E. coli can suppress inflammation via the mammalian melanocortin-1 receptor (MC1R): possible endocrine-like function for microbes of the gut. NPJ Biofilms Microbiomes 2017;3:31. ArticlePubMedPMCPDF
- 15. Ito Y, Betsuyaku T, Nasuhara Y, Nishimura M. Lipopolysaccharide-induced neutrophilic inflammation in the lungs differs with age. Exp Lung Res 2007;33:375-84.ArticlePubMedPMC
- 16. Kitamura Y, Hashimoto S, Mizuta N, Kobayashi A, Kooguchi K, Fujiwara I, et al. Fas/FasL-dependent apoptosis of alveolar cells after lipopolysaccharide-induced lung injury in mice. Am J Respir Crit Care Med 2001;163:762-9.ArticlePubMed
- 17. Ignar DM, Andrews JL, Jansen M, Eilert MM, Pink HM, Lin P, et al. Regulation of TNF-alpha secretion by a specific melanocortin-1 receptor peptide agonist. Peptides 2003;24:709-16.ArticlePubMed
- 18. Montero-Melendez T, Gobbetti T, Cooray SN, Jonassen TE, Perretti M. Biased agonism as a novel strategy to harness the proresolving properties of melanocortin receptors without eliciting melanogenic effects. J Immunol 2015;194:3381-8.ArticlePubMed
- 19. Szarka RJ, Wang N, Gordon L, Nation PN, Smith RH. A murine model of pulmonary damage induced by lipopolysaccharide via intranasal instillation. J Immunol Methods 1997;202:49-57.ArticlePubMed
- 20. Patel BV, Wilson MR, O’Dea KP, Takata M. TNF-induced death signaling triggers alveolar epithelial dysfunction in acute lung injury. J Immunol 2013;190:4274-82.ArticlePubMedPMC
- 21. Star RA, Rajora N, Huang J, Stock RC, Catania A, Lipton JM. Evidence of autocrine modulation of macrophage nitric oxide synthase by alpha-melanocyte-stimulating hormone. Proc Natl Acad Sci U S A 1995;92:8016-20.ArticlePubMedPMC
- 22. Catania A, Rajora N, Capsoni F, Minonzio F, Star RA, Lipton JM. The neuropeptide alpha-MSH has specific receptors on neutrophils and reduces chemotaxis in vitro. Peptides 1996;17:675-9.ArticlePubMed
- 23. Gonindard C, Goigoux C, Hollande E, D’Hinterland LD. The administration of an alpha-MSH analogue reduces the serum release of IL-1 alpha and TNF alpha induced by the injection of a sublethal dose of lipopolysaccharides in the BALB/c mouse. Pigment Cell Res 1996;9:148-53.ArticlePubMed
- 24. Bhardwaj RS, Schwarz A, Becher E, Mahnke K, Aragane Y, Schwarz T, et al. Pro-opiomelanocortin-derived peptides induce IL-10 production in human monocytes. J Immunol 1996;156:2517-21.ArticlePubMedPDF
Citations
Citations to this article as recorded by

- Exosomes Derived from ADSCs Attenuate Sepsis-Induced Lung Injury by Delivery of Circ-Fryl and Regulation of the miR-490-3p/SIRT3 Pathway
Weijun Shen, Xuan Zhao, Shitong Li
Inflammation.2022; 45(1): 331. CrossRef - Dichotomous Role of Tumor Necrosis Factor in Pulmonary Barrier Function and Alveolar Fluid Clearance
Rudolf Lucas, Yalda Hadizamani, Perenlei Enkhbaatar, Gabor Csanyi, Robert W. Caldwell, Harald Hundsberger, Supriya Sridhar, Alice Ann Lever, Martina Hudel, Dipankar Ash, Masuko Ushio-Fukai, Tohru Fukai, Trinad Chakraborty, Alexander Verin, Douglas C. Eato
Frontiers in Physiology.2022;[Epub] CrossRef - NDP-MSH treatment recovers marginal lungs during ex vivo lung perfusion (EVLP)
Caterina Lonati, Michele Battistin, Daniele E. Dondossola, Giulia A. Bassani, Daniela Brambilla, Riccardo Merighi, Patrizia Leonardi, Andrea Carlin, Marica Meroni, Alberto Zanella, Anna Catania, Stefano Gatti
Peptides.2021; 141: 170552. CrossRef - Antifibrotic and Anti-Inflammatory Actions of α-Melanocytic Hormone: New Roles for an Old Player
Roshan Dinparastisaleh, Mehdi Mirsaeidi
Pharmaceuticals.2021; 14(1): 45. CrossRef - Activation of Melanocortin Receptors as a Potential Strategy to Reduce Local and Systemic Reactions Induced by Respiratory Viruses
Caterina Lonati, Stefano Gatti, Anna Catania
Frontiers in Endocrinology.2020;[Epub] CrossRef

 , Jin-Young Kim1
, Jin-Young Kim1

 PubReader
PubReader ePub Link
ePub Link Cite
Cite