Brain-lung interaction: a vicious cycle in traumatic brain injury
Article information

Abstract
The brain-lung interaction can seriously affect patients with traumatic brain injury, triggering a vicious cycle that worsens patient prognosis. Although the mechanisms of the interaction are not fully elucidated, several hypotheses, notably the “blast injury” theory or “double hit” model, have been proposed and constitute the basis of its development and progression. The brain and lungs strongly interact via complex pathways from the brain to the lungs but also from the lungs to the brain. The main pulmonary disorders that occur after brain injuries are neurogenic pulmonary edema, acute respiratory distress syndrome, and ventilator-associated pneumonia, and the principal brain disorders after lung injuries include brain hypoxia and intracranial hypertension. All of these conditions are key considerations for management therapies after traumatic brain injury and need exceptional case-by-case monitoring to avoid neurological or pulmonary complications. This review aims to describe the history, pathophysiology, risk factors, characteristics, and complications of brain-lung and lung-brain interactions and the impact of different old and recent modalities of treatment in the context of traumatic brain injury.
INTRODUCTION
Traumatic brain injury (TBI) is one of the leading causes of death and disability worldwide, with enormous economic and social consequences [1]. TBI results from direct damage to the brain caused by an external, physical force, which can lead to a state of decreased consciousness, physical functioning, or deterioration of cognitive ability [2]. Many secondary lesions in patients with TBI occur in a medium-long term period, and the appearance of brain and pulmonary complications is highly prevalent. Direct brain injury and altered levels of consciousness decrease the protection of the airways and alter the natural defense barrier, which adds to reduced mobility and multiple pathophysiological deficits inherent to the injury [3] and triggers several lung injuries, like neurogenic pulmonary edema (NPE), acute respiratory distress syndrome (ARDS), and ventilation-associated pneumonia (VAP). In the same way, lung injuries can affect the brain due to alterations in pulmonary physiology having repercussions at the systemic level, leading to neurological disorders which can be triggered mainly by hypoxia and intracranial hypertension [3,4].
The brain and lungs share fundamental connections that are compromised in patients with TBI. This injury becomes a vicious cycle that worsens patient condition (Figure 1). Considering the increasing global incidence of TBI, deficient access to healthcare in many parts of the world, and inadequate methods of treatment [1], we present a narrative review of the available evidence on the mechanisms of brain-lung interaction in patients with trauma as well as new therapeutic implications and possible management strategies that could reduce brain and pulmonary complications and improve the prognosis of patients in order to provide a basis for future research.
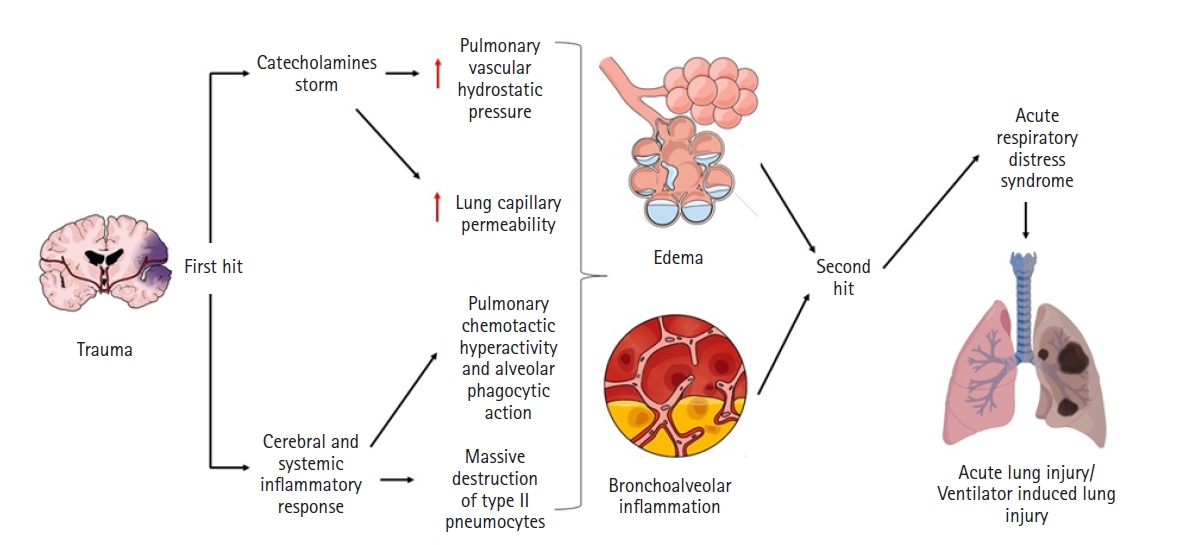
Vicious circle of the interaction between acute lung injury in patients with traumatic brain injury: a double impact model.
HISTORY
Extracranial complications frequently occur in patients with TBI; among them, pulmonary disorders are common and deteriorate patient condition, leading to serious neurological outcomes. Brown-Séquard [5] was the first to describe the interaction between TBI and the lungs in 1871 in his experimental traumatic injury to the pons of guinea pigs that resulted in pulmonary hemorrhage and edema. In 1969, Simmons et al. [6] evaluated an autopsy series of patients who died from TBI that reported the vast majority of those who died within few minutes after trauma to have pulmonary edema, which allowed them to affirm that TBI is decisive in the development of pulmonary edema with consequent respiratory dysfunction. In 1976, Theodore and Robin introduced the "explosion theory" to explain NPE in patients with TBI [7] that declared an excess of catecholamines and consequent positive regulation of sympathetic signal transduction to be responsible for the increase in pulmonary venous pressure and transudative edema. Finally, although NPE was estimated to have an incidence of only 1% after cerebral trauma in 1997, a mortality of 60%–100% was demonstrated regardless of its etiology, which triggered the alarms to consider it a life-threatening condition after it was demonstrated to increase of intracranial pressure (ICP), where immediate therapeutic interventions are essential [8].
Likewise, in terms of frequent pulmonary complications, ARDS is also notable. Its concept has evolved from the old name idiopathic pulmonary anasarca, postulated by Laënnec in 1821, until its recent definition in Berlin [9]. In 1967, Ashbaugh et al. [10] presented the concept of ARDS as respiratory distress syndrome, which was improved in 1988 by Murray et al. [11] by including the description of a multi-section system. Later, the European-American Consensus Conference conceptualized the criteria for acute lung injury (ALI). However, it was decided not to use the ALI term and to stratify ARDS into three levels of severity in the more recent Berlin definition [9].
In 1997, Bratton and Davis [12] evaluated the incidence of ALI in comatose patients after isolated TBI and found that it was 20%, showing a three-fold greater probability of dying or surviving in a vegetative state. In a study published in 2003, it was reported that 31% of patients with severe TBI developed ALI, with a greater number of days in mechanical ventilation (MV), a worse neurological outcome, and mortality of 38% [13]. The presence of ALI or ARDS complicates the treatment of patients with TBI because hypoxia causes additional damage to the brain and because therapies used to protect the lungs and improve patient oxygenation can reduce cerebral blood flow and increase ICP [14]. In a 20-year retrospective cohort study of patients with TBI, an increase in the prevalence of ALI or ARDS was found, and a greater association with comorbidities, such as congestive heart failure, hypertension, chronic obstructive pulmonary disease, chronic kidney disease, and liver failure, as well as increased risk of sepsis, multi-organ dysfunction, and in-hospital mortality was found [15].
There is a high frequency of infection in patients with TBI due to a predisposition attributed to compromised host defenses after brain trauma and the need for MV in these patients [16]. In 1992, Piek et al. [17] evaluated the determinants of recovery after severe brain trauma and found that pneumonia was the second most frequent complication (40.6%) on the fifth to tenth days after the trauma; furthermore, they emphasized the fact that many of them were not preventable in critically ill patients and could only be managed through quick diagnosis, prompt etiology identification, and opportune treatment [17]. Later, pneumonia was found in 60% of 125 patients with closed head trauma. Of which, 47.8% corresponded to early pneumonia and were associated with lower scores on the Glasgow coma scale (GCS), longer intubation time, intensive care duration, and hospitalization [18].
VAP is among the most important subtypes of nosocomial infections, and the incidence of this type of pneumonia in patients with brain injury ranges from 28% to 40% [19]. In 1999, Ranieri et al. [20] demonstrated for the first time that MV induced a cytokine response in the lungs and plasma, which was associated with higher rates of multi-organ failure. However, they suggested that attenuation of this inflammatory response could be a useful strategy to minimize overstretching and cell recruitment in lungs, with possible improvements in clinical outcomes. In 2004, Bronchard et al. [21] published a prospective observational study of patients with TBI who required tracheal intubation for neurological reasons and ventilation for at least 2 days and reported a 41.3% incidence of early-onset pneumonia with low arterial oxygen pressure (PaO2), decreased inspired fraction of oxygen (FiO2), fever, hypotension, and intracranial hypertension. They concluded that early-onset pneumonia led to secondary lesions and neurological deterioration.
BRAIN-LUNG INTERACTIONS IN BRAIN TRAUMA
From Brain to Lung
Physiopathology
Shortly after presenting brain damage of any kind, preclinical lung injury appears even without obvious symptoms–this effect manifests with alterations in the mechanics of the respiratory supply of oxygen and the elimination ofsystem and hypoxemia [22]. Although the mechanisms by which brain damage leads to alterations in lung function are not clear, the development of NPE, inflammatory processes, neurotransmitter-related compromise, and even adverse effects secondary to the administration of neuroprotective therapies can be considered fundamental [23]. A well-recognized theory to explain the pathophysiology responsible for lung damage is “blast injury,” which suggests that a sympathetic storm after a sudden increase in ICP induces a massive release of catecholamines. This results in a transient increase in intravascular pressure, rupture of the alveolar-capillary membrane, and consequent development of NPE [22].
As well as NPE constituting a fundamental part of the genesis of pulmonary dysfunction after TBI, the systemic inflammatory response also plays an important role in its development [24]. The intracranial inflammatory response occurs after brain injury, and pro-inflammatory cytokines, such as interleukins (IL-1, IL-6, IL-8) and tumor necrosis factor, are produced locally in the injured brain tissue. First, microglia and astrocytes are the main sources of inflammatory mediators. Then, the alteration of blood-brain barrier permeability allows its discharge into systemic circulation. Likewise, all this production of inflammatory mediators ("first hit") concomitant to the procedures used in the treatment of patients with TBI; such as MV; surgical procedures; or complications, namely infections ("second hit"), make up the recently described “double hit” model (Figure 1) [4].
Neurogenic pulmonary edema
NPE is a clinical condition that manifests as acute respiratory distress, occurring secondary to insult to the central nervous system (CNS). By definition, this condition is characterized by a large accumulation of extravascular pulmonary fluid, which appears suddenly and immediately after serious lesions of the CNS, mostly the brainstem [25]. If the clinical presentation is evident, the diagnosis should be assumed when acute pulmonary edema is observed in association with CNS injury in the absence of primary pulmonary or cardiovascular injury. However, this idea is imprecise because the literature does not present a complete understanding of the exact pathogenesis [26-28].
Nowadays, it is known that alteration of the CNS causes a sympathetic overflow, leading to a state of systemic vasoconstriction. This leads to the accumulation of blood from systemic circulation into the pulmonary circulation, resulting in pulmonary hypertension and an increase in pulmonary capillary hydrostatic pressure [29]. The change in pressures is responsible for the leakage of intravascular fluid to both the alveoli and the pulmonary interstitial space through two mechanisms: first, Starling forces, and second, changes in permeability in the capillary walls [25].
Elevated systemic arterial pressure and increased left atrial and pulmonary hydrostatic pressure, culminating in pulmonary edema, are attributed to peaks in sympathetic activity, particularly α-adrenergic, induced by lesions of the hypothalamus or spinal cord [30]. This sympathetic hyperactivity is triggered by a sudden increase in ICP or a decrease in cerebral blood flow [31].
Acute respiratory distress
In patients with brain lesions, a high incidence rate of ARDS has been described [4]. This affects the morbidity and mortality of patients with brain lesions in all cases, increasing inpatient mortality until 3 times when it appears after TBI. Risk factors for the development of ARDS have been identified. First, the severity of the initial brain injury characterized by a low GCS and abnormalities in initial brain computed tomography (CT) (displacement midline and global CT findings) and second, induced hypertension, administration of vasoactive drugs, and a history of drug abuse have been reported to be independent factors of ARDS in severe TBI. Similarly, there are general risk factors, such as male sex; young age; ethnicity; and a history of chronic arterial hypertension, diabetes, chronic obstructive pulmonary disease, development of sepsis, cardiovascular, renal, and hematological dysfunctions [15,32]. On the other hand, it has been determined over the years that the distribution of ARDS has a bimodal character, consisting of an early peak on the second or third day after the start of MV and a later peak on the eighth or ninth day after the start of ventilation, often related to pneumonia [4].
Ventilation-associated pneumonia
VAP is defined as an infection of the lung parenchyma that occurs in patients exposed to invasive MV for at least 48 hours. Currently, this pathology remains one of the most frequent infections in those patients who require invasive MV, and despite recent advances in microbiological tools and epidemiology, the diagnostic criteria remain controversial [33]. MV is an effective life-saving intervention method for critically ill patients. Thus, it is widely used in intensive care units (ICUs). This strategy allows the supply of oxygen and the elimination of carbon dioxide (CO2) with strict control of PaO2 and PaCO2. The main objective of MV is the prevention of secondary cerebral ischemia and incrementally positive neurological outcomes. However, prolonged MV can increase the risk of infection and a variety of complications [4].
Ventilation time and ICU stay have been reported to be significantly longer in patients with VAP than in those without VAP. An Egyptian study found that the incidence of VAP increased from 5% in patients who received 1 day of MV to 65% in patients who received 30 days of MV [34]. An artificial airway must be established to perform MV, which changes the defense function of the resident airway mucosa and decreases the ability to swallow and the ability of cilia to clear. Subsequently, bacteria can enter the lower respiratory tract directly or cross the space between the wall of the tracheal tube and the airways to begin the infectious process [35].
VAP has been frequently identified in neurological patients due to decreased level of consciousness, micro aspirations, and even massive aspiration. The main risk factors for developing VAP in patients with brain injury are polytransfusion, age, obesity, diabetes, immunosuppression status, chronic lung disease, and the use of barbiturates. In addition, sedation, the use of muscle relaxants, prior antibiotic treatment, and the absence of an inclined position during MV increase the risk of developing VAP [4]. A study conducted in Serbia in 2015 showed that the incidence of VAP in patients with severe TBI was up to 49.7%, much higher than the mean incidence of VAP without TBI [36].
The microorganisms that cause VAP vary with respect to the duration of MV; the length of stay in the ICU; the previous, prolonged or irresponsible use of antimicrobials; the local microbial prevalence; and the appearance of any potential epidemic phenomenon in a given ICU. The most common gram-negative bacteria involved in VAP are Pseudomonas aeruginosa, Escherichia coli, Klebsiella pneumoniae, and Acinetobacter species, while Staphylococcus aureus is the main gram-positive microorganism, being the most common pathogen reported in VAP in patients with severe TBI [37,38].
In general, it is recognized that a normal oropharyngeal flora is generally involved in VAP that occurs in the first 4 days of hospitalization in previously healthy patients who have not received or receive antibiotics regularly or inadequately. On the other hand, in patients who present the clinical characteristics of VAP after at least 5 days of hospitalization and who present its risk factors, it is more likely that their infection is due to multidrug-resistant (MDR) pathogens. However, MDR pathogens can be isolated in early-onset VAP, primarily in the presence of certain risk factors, such as antimicrobial exposure within the previous 90 days [39,40].
From the Lung to the Brain
Physiopathology
Due to a large number of communication pathways between the peripheral nervous system and the CNS [9], lung injuries or pathologies that affect proper oxygenation can lead to alterations at the neuronal level, like memory problems, disorientation, cognition, and language alterations, through various routes [41]. Among these pathways, we find three to be fundamental: humoral, neural, and cellular. The first pathway consists of the recruitment of monocytes or macrophages in the lungs, which increases the levels of inflammatory mediators that can directly reach the CNS through the humoral route. The neuronal pathway refers to the fact that there is a connection between the brain and the nucleus of the solitary tract through the afferents of the vagal pathway. Finally, the cellular pathway is directly regulated by the release of tumor necrosis factor in the lungs that stimulates the release of monocyte chemoattractant protein-1 at the brain level, which increases the recruitment of activated monocytes both at the level of the CNS and the periphery nervous system [9].
Cerebral hypoxia
Hypoxemia is a potential risk factor for the development of long-term cognitive impairment. Lower PaO2 values were associated with cognitive impairment in general and executive dysfunction specifically. Long-term neuropsychological damage is common in survivors of ALI [42]. Despite the mechanism of connection or integration that causes neurological alterations not being widely understood, several experimental studies have tried to explain the mechanism by which lung damage can lead to neurological disorders and uncover the role of hypoxemia in these changes. In a 2005 experimental study with 14 pigs, Fries et al. [43] induced hypoxemia in two groups by repeated lung lavages (lung injury group) or by reducing the inspired oxygen fraction (hypoxia-only group), concluding that the same degree of hypoxemia induced in the lavage model of ALI results in greater brain damage, assessed by S-100 protein and histopathologic findings when compared to the group in which hypoxemia was induced at the same degree by reducing the inspired oxygen fraction. This suggests that ALI leads to neuropathological changes independent of hypoxemia [43].
Bickenbach et al. [44] also examined the influence of ALI and hypoxia on neurological outcomes in pigs. In their study, they demonstrated that neurocognitive compromise after ALI seems due to a more pronounced inflammatory response and complex MV. However, these changes only resulted in mild changes in histopathology in terms of perivascular inflammation in the hippocampal CA1 sector, and the caudate nucleus and the putamen were not significantly different between the groups [44].
Mechanisms for the deterioration of neurologic function after lung injury remain unclear and may include hypoxia and the effects of MV and its consequential inflammatory response [44]. A systematic review in 2021 showed an association between MV and acute cognitive impairment, and preclinical papers showed acute cognitive impairment after MV, describing greater neuroinflammation and lower cognitive scores in subjects with a longer MV duration [45]. On the other hand, patients with ARDS may experience long periods of hypoxia, leading to the hypothesis that they might develop brain lesions and atrophy similar to those observed in other disorders with concomitant hypoxia [46]. Kamuf et al. [47] carried out the first study focusing on the neuroinflammatory injury potential of early ARDS, and they found hints indicating an increased cerebral expression of inflammatory cytokines within 6 hours after the onset of lung injury but no signs of histopathologic injury [47]. The mechanism of changes in the brain-lung relationship that occur in hypoxic situations could be explained by specific signaling pathways. Some of these pathways have been established in animal models, such as in naked mole-rat hypoxia models, in which hypoxia altered the Akt/mammalian target of rapamycin pathway, which correlated with a general decrease in the ribosomal protein biosynthesis machinery activity in the lungs and brain [48].
Intracranial hypertension
In brain-injured patients, especially those in critical care, there is a serious detriment to respiratory function with the elevation of PaCO2. This directly causes the vasodilation of cerebral arteries and a consequent increase in cerebral blood volume, which might result in a higher ICP if intracranial compliance is reduced [22]. Moreover, PaO2 and PaCO2 can also change with MV and positive end-expiratory pressure (PEEP), which raise ICP by increasing pleural pressure and diminishing venous return. [49]. Intracranial hypertension and ARDS were thought to be independent pathological entities, but recent results have shown that they both interact and actually trigger each other. A study in mechanically ventilated pigs found that ARDS induces a systemic inflammatory response with aberrantly elevated cytokines and neutrophil counts, which has a detrimental effect on the brain and acts synergistically with intracranial hypertension [50].
Therapeutic Implications: the Conflict between the Brain and Lungs
The management of patients with TBI represents a great challenge and includes multiple strategies that seek to preserve the brain-lung balance. The objective of ventilatory support is to maintain adequate oxygenation (PaO2 ≥60 mm Hg) and avoid hypercapnia [51] in addition to protecting ICP and thus preventing secondary brain injury [22,52]. For this reason, the traditional ventilatory strategy for the prevention and treatment of intracranial hypertension was to impart high tidal volume to maintain mild hypocapnia (PaCO2 –30 to 35 mm Hg) [51]. However, it has been shown that high tidal volume can affect the brain, inducing intracranial hypertension and causing a deleterious event (second hit) in the lung, which is a particularly sensitive organ [22].
A neurological study of ICU patients established that high tidal volume ventilation was an independent predictor of ARDS in patients with severe brain injury [53]. Likewise, a study in rats found that low tidal volume minimized lung morphological and functional changes, improving oxygenation and reducing lung damage compared to the use of high tidal volumes [54]. Thus, research suggests that maintaining a low tidal volume could be more beneficial in patients with or without ARDS since it reduces the risk of pulmonary complications after TBI [55].
According to the guidelines of the “Brain Trauma Foundation,” prolonged prophylactic hyperventilation with PaCO2 ≤25 mm Hg is not recommended for the reduction of elevated ICP, and it should be avoided, especially during the first 24 hours after the injury when the cerebral blood flow often declines critically. In addition, it is mentioned that hyperventilation with measurements of jugular oxygen saturation or partial pressure of oxygen of brain tissue to monitor oxygen supply is only temporarily recommended [56]. These recommendations are based on the fact that hyperventilation can be harmful since severe hypocapnia and subsequent cerebral vasoconstriction can lead to hypoxia in brain tissue and compromised blood flow velocities and compliance. Finally, when ARDS and TBI coexist, it is necessary to find a balance between CO2 control and lung protection, although there are no absolute contraindications for the use of protective ventilation in the treatment of TBI without significant intracranial hypertension. Thus, PaCO2 values should be established on a case-by-case basis in accordance with ICP [51,52].
On the other hand, PEEP increases pleural and intrathoracic pressure and, consequently, can alter central venous return and cause an increase in ICP; therefore, the management of null or low PEEP (≤5 cm H2O) has been recommended in mechanically ventilated brain-injured patients [57]. However, the transmission of PEEP into the thoracic cavity depends on the properties of the lungs and chest wall. An experimental study conducted by Chapin et al. [58] showed that when the lung elastance is high, PEEP can minimize airway pressure transmission, whereas high elastance in the chest wall can significantly increase pleural pressure and eventually ICP. Both ideas were supported by several studies. First, a prospective study of 21 comatose patients with severe head injury or subarachnoid hemorrhage who required MV and PEEP demonstrated that PEEP exerts no significant effects on cerebral hemodynamics in patients with high respiratory system elastance [49]. Likewise, in a prospective pilot study that evaluated 20 patients with TBI and ARDS, it was reported that an increase in PEEP up to 15 cm H2O resulted in an increase in oxygen pressure and saturation in brain tissue without an increase in ICP or a decrease in cerebral perfusion pressure (CPP) [59]. Similarly, a study found that the effect of PEEP on ICP was more profound in patients with higher elastance chest walls [60]. Nevertheless, PEEP can alter CPP in hypovolemic conditions [57], which seems to indicate that an increase in PEEP can be safely applied to impart beneficial brain effects in brain-injured ARDS patients as long as they are normovolemic with high lung elastance and low chest wall elastance.
Recruitment maneuvers (RMs) have been reported as useful strategies capable of improving oxygenation, restoring alveolar recruitment, and optimizing ventilation-perfusion mismatch [52]. However, for the same reasons as PEEP, RMs could lower blood pressure and increase ICP by interfering with the return of venous blood, causing an elevation in intrathoracic pressure. Thus, the use of RMs can be safe only with strict monitoring of systemic and cerebral parameters and the use of progressive maneuvers [4].
Likewise, the prone position has been shown to increase oxygenation through different mechanisms, such as net recruitment, increased homogeneous distribution of alveolar ventilation, and protection from ventilation-associated lung injury [4]. The clinical case of a patient with thoracic and brain trauma who subsequently developed ARDS showed an improvement in lung function indices with moderate and very transient effects of ICP that resolved shortly after the change in position (supine position) [61]. Nevertheless, this maneuver has been poorly studied in patients with brain injuries, so prone positioning may be considered in patients with concurrent severe ARDS (PaO2/FiO2 ratio <150) and TBI but do not have significant ICP elevation [62].
It has recently been shown that inflammasomes NLRP 1,2,3 (NOD-like receptors purine domain-containing) NLRC 4 (Nod-like receptor CARD domain-containing), and AIM 2 (absent in melanoma 2) play a role in TBI, facilitating the rapid activation of the innate immune response [63], which leads to exaggerated systemic inflammation and contributes to the damage of other organs, favoring the pathogenesis of ALI/ARDS after TBI [64]. Therefore, these inflammasome proteins can serve as possible biomarkers to assess the severity of injury and as possible therapeutic targets for inhibition [63].
Currently, there are no effective therapies to mitigate disability. However, new studies are paving the way for future interventions. In a study by Lee et al. [65], they found evidence that inhibiting neuronal death and preventing the activation of the inflammasome provides beneficial effects by reducing the potentially harmful consequences of activated microglia and leukocyte infiltration after penetrating TBI [65]. Also, inflammasome inhibition through the use of neutralizing antibodies against inflammasome proteins has been tested and has had some success in preclinical models of TBI [63].
In a study evaluating the effects of enoxaparin after TBI, a significant decrease in the ALI score as well as neutrophil and macrophage infiltration into the lungs 24 hours after injury was found. This study demonstrated that enoxaparin attenuates ALI and inhibits the expression of the inflammasome in the brain and lungs after TBI, supporting the hypothesis that the inhibition of the neural-respiratory inflammasome axis that is activated after TBI may have therapeutic potential [66].
Regarding NPE patients, few studies have reported on specific treatments in humans, and only a few studies in animals have focused on treatment with α-blockers to limit massive sympathetic discharge after brain injuries [4]. This emphasizes the need for more studies with directed strategies to reduce the organic consequences and improve patient prognosis.
CONCLUSION
The brain-lung relationship arises from the interaction of several factors which are triggered by trauma responses and can communicate between the organs through different pathways. Nevertheless, almost always after a TBI, only the brain effect at the pulmonary level is considered, with very little consideration of the detrimental reverse consequences. Acute lung damage caused by ARDS or MV can worsen neurological functions in patients with TBI by humoral, neuronal, and cellular communications and by the interaction of numerous elements released locally or systemically in trauma response. Hence, therapeutic strategies should be able to minimize the impact of TBI in critical patients, both at the pulmonary and neurological levels.
MV can have a beneficial effect on oxygenation and cerebral perfusion in patients with TBI. However, the estimation of tidal volume, PEEP, and RMs is also necessary in addition to considering systemic and cerebral parameters, such as ICP, PaCO2, PaO2, and properties of the lungs and chest wall due to the close relationship that they have to the elevation of ICP. Finally, we conclude that the management of patients with TBI should be established on a case-by-case basis considering individual characteristics to avoid the appearance of neurological and pulmonary complications that influence prognosis or constitute long-term sequelae with greater morbidity and mortality, additional costs to health services, and the inevitable decrease in patient quality of life. Although new strategies targeting the inflammatory cascade are being investigated, experimental and clinical studies are needed to evaluate the brain and lung level effects.
KEY MESSAGES
• The brain-lung interaction is key in the pathophysiology of complications in traumatic brain injury.
• Neurogenic pulmonary edema, acute respiratory distress, and ventilator-associated pneumonia are the main conditions passed from the brain to the lungs.
• Cerebral hypoxia and intracranial hypertension are the main pulmonary-to-brain conditions.
Notes
CONFLICT OF INTEREST
No potential conflict of interest relevant to this article was reported.
AUTHOR CONTRIBUTIONS
Conceptualization: all authors. Data curation: AACA, EADV, JAAC, IDLM, MPBR. Formal analysis: AACA, EADV, JAAC, IDLM, MPBR. Methodology: all authors. Project administration: IDLM, LRMS, PG, TJ. Writing–original draft: all authors. Writing–review & editing: all authors.