Abstract
-
Background:
- Flecainide is an antiarrhythmic agent that is used primarily in the treatment of cardiac arrhythmias. Some evidences also suggest that flecainide can participate in alveolar fluid clearance and inflammatory responses. This experiment was aimed to evaluate the effects of flecainide on sepsis induced acute lung injury in a rat model.
-
Methods:
- Rats were treated with subcutaneous infusion of saline or flecainide (0.1 or 0.2 mg/kg/hr) by a mini-osmotic pump. Subcutaneous infusion was started 3 hours before and continued until 8 hours after intraperitoneal injection of saline or endotoxin. Animals were sacrificed for analyses of severity of acute lung injury with wet to dry (W/D) ratio and lung injury score (LIS) in lung and inflammatory responses with level of leukocyte, polymorphonuclear neutrophils (PMNs) and inteleukin-8 (IL-8) in bronchoalveolar lavages fluid (BALF).
-
Results:
- Flecainide markedly improved dose dependently sepsis induced acute lung injury as analysed by W/D ratio (from 2.24 ± 0.11 to 1.76 ± 0.09, p < 0.05) and LIS (from 3 to 1, p < 0.05), and inflammatory response as determined by leukocyte (from 443 ± 127 to 229 ± 95, p < 0.05), PMNs (from 41.43 ± 17.63 to 2.43 ± 2.61, p < 0.05) and IL-8 (from 95.00 ± 15.28 to 40.00 ± 10.21, p < 0.05) in BALF.
-
Conclusions:
- Flecanide improve sepsis induced acute lung injury in rats by controlling inflammatory responses.
-
Keywords: lung injury; cytokine; flecainide; leukocyte; sepsis
Introduction
Acute respiratory distress syndrome (ARDS) is a complex syndrome that characterize as severe pulmonary inflammatory responses with high morbidity and mortality. Although the exact pathogenesis for ARDS is not yet fully defined. But sepsis-induced over-activation of inflammatory cells involving macrophages and neutrophils is one of variable reasons to play critical roles in the development of ARDS.[1] Thus, nontoxic molecules that regulate inflammation may provide an innovative therapeutic strategy.
Flecainide, a strong blocker of sodium channel, has favorable electrophysiologic effects in the management of cardiac arrhythmia. So it has been widely used in clinical practice. In addition, some reports suggest that flecainide can participate to regulate lung fluid homeostasis[2] and neutrophil action.[3] Thereby flecainide might also participate in modulating alveolar fluid clearance (AFC) and cellular recruitment during acute inflammatory processes. Therefore, we proposed the hypothesis that the efficacy of flecainide include not only antiarrhythmic effect but also anti-inflammatory effect in sepsis induced acute lung injury (ALI).
In the present experiments, we evaluated whether flecainide improves sepsis induced acute lung injury as determined by wet to dry (W/D) ratio and lung injury score (LIS) in lung and inflammatory responses involving level of leukocyte, polymorphonuclear neutrophils (PMNs) and inteleukin-8 (IL-8) in bronchoalveolar lavages fluid (BALF) in a rat model.
Materials and Methods
1) Materials and animals
Flecainide was obtained from Hana pharm (Seoul, Korea). Sevoflurane was obtained from Abbott Laboratories (Chicago, IL, USA). Escherichia coli 0111:B4 endotoxin was bought from Sigma-Aldrich (St. Louis, MO, USA). Immunoreactive IL-8 were quantified using commercially available enzyme-linked immunosorbent assay kits (R & D Systems, Minneapolis, MN, USA), in accordance with manufacturer’s instructions and as described previously. [4] Male SD rats, 8–12 weeks of age, were purchased from Damul science (Daejeon, Korea). The animals were kept on a 12hours light/dark cycle with free access to water and food. All experiments were conducted according to institutional review board approved protocols.
2) Animal model of endotoxin-induced acute lung injury
Animals were randomly attached to one of five groups involving rats receiving subcutaneous (SC) saline and intraperitoneal (IP) saline (S-S group, n = 14), those receiving SC saline and IP endotoxin (25 mg/kg) (S-E group, n = 14), those receiving SC flecainide (0.2 mg/kg/h) and IP saline (F-S group, n = 14), those receiving SC flecainide (0.1 mg/kg/h) and IP endotoxin (F 0.1-E group, n =14), those receiving SC flecainide (0.2 mg/kg/h) and IP endotoxin (F 0.2-E group, n = 14). Animals were initially anesthetized with sevoflurane. After the swabbing on the interscapular area, a mini-osmotic pump (model 2001; Alzet, Alza, Palp Alto, CA, USA) was implanted to administer saline or flecainide at a rate of 0.1 or 0.2 mg/kg/h for a period of 11 hours totally. SC infusion of saline or flecainide using by a mini-osmotic pump was started 3 hours before and continued until 8 hours after injection of saline or endotoxin when all animals were killed. ALI was induced by IP injection of 25 mg/kg Escherichia coli 0111:B4 endotoxin into animals. With this model, ALI, as characterized by neutrophil infiltration into the lung interstitium, progress of interstitial edema, and increment of proinflammatory cytokine occurs after injection of endotoxin, with the greatest accumulation of neutrophils into the airways and histopathologic injury being present 8 hours after endotoxin exposure.[5-7] Animals were assigned to 14 rats per each group for the assessment of W/D ratio and histopathologic examination of the lung (n = 7) and analysis of BALF (n = 7), respectively. Animals were euthanized to collect BALF and lung tissues at 8 hours after the IP saline or endotoxin. At this time point, the number of white blood cell, percentages of PMNs and the concentration of IL-8 in BALF, W/D ratio and histopathologic examination were evaluated.
3) W/D ratio
All animals used for W/D ratios were of identical ages. The left lobes were excised, rinsed briefly in phosphate buffered saline, blotted, then weighed to acquire the wet weight. Next, lungs were dried in an oven at 70°C for 7 days to acquire the dry weight.
4) Histopathologic examination
The right lobes were fixed by instillation of 10% glutaraldehyde solution through the right main bronchus at 20cmH2O. The lungs were embedded in paraffin, and the sections were stained with hematoxylin and eosin. Lung injury was scored as follows by a pathologist (who was unaware of the nature of the experiment) under light microscopy as previously described[8]—0 (no reaction in alveolar walls), 1 (diffuse reaction in alveolar walls, primarily neutrophilic, no thickening of alveolar walls), 2 (diffuse presence of inflammatory cells [neutrophilic and mononuclear] in alveolar walls and slight thickening), 3 (distinct thickening [two or three times] of the alveolar walls due to the presence of inflammatory cells), 4 (alveolar wall thickening with up to 25% of the lung consolidated), 5 (alveolar wall thickening with more than 50% of the lung consolidated). To count the number of neutrophils in the airspace, five randomly selected fields per slide were read at ×400 magnification by the pathologist. Fields containing large vessels or bronchi were excluded. The number of neutrophils per field was normalized to alveoli per field to control for inflation of the lung.
5) BALF
Bronchoalveolar lavages were obtained by cannulating the trachea with a blunt 16-gauge needle and then lavaging the lungs three times with 10 mL of iced phosphate buffered saline. The BALF was analyzed for cell count and cell differentiation. For cell differentiation, a cytocentrifuged spin preparation (CF-RD, Sakura, Tokyo, Japan) of the BALF was stained with Wright. Using a cell counter (XE2100, Sysmex, Kobe, Japan), the numbers of leukocytes in BALF were counted. The fluid was centrifuged at 250 g at 4ºC for 20 min to remove the cells. The cell-free supernatant was divided into several aliquots and stored at -80ºC for measurements of IL-8.
6) Statistical analysis
SPSS (Windows ver. 21.0, SPSS Inc., Chicago, IL, USA) was used for the statistical analysis. Data are presented as mean ± standard error of the mean except the lung injury score which is given as a median for each experimental group. Data from three or more groups were compared using one-way analysis of variance followed by the Scheffé multiple comparison test. Pairwise comparisons were made with the Student’s t test. A value of p < 0.05 was considered significant.
Results
1) W/D ratios
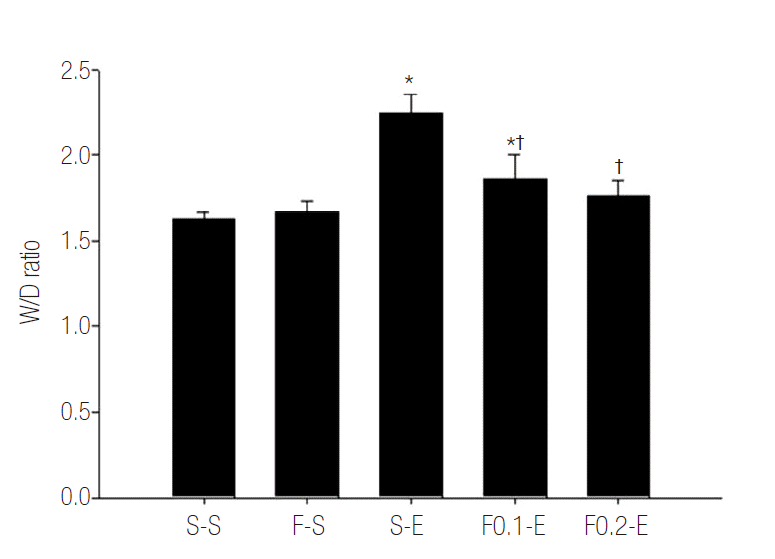
W/D ratio was increased in S-E group compared with in S-S group. Flecainide reduced the degree of increase in these ratio (Table 1, Fig. 1).
2) Histopathologic grading
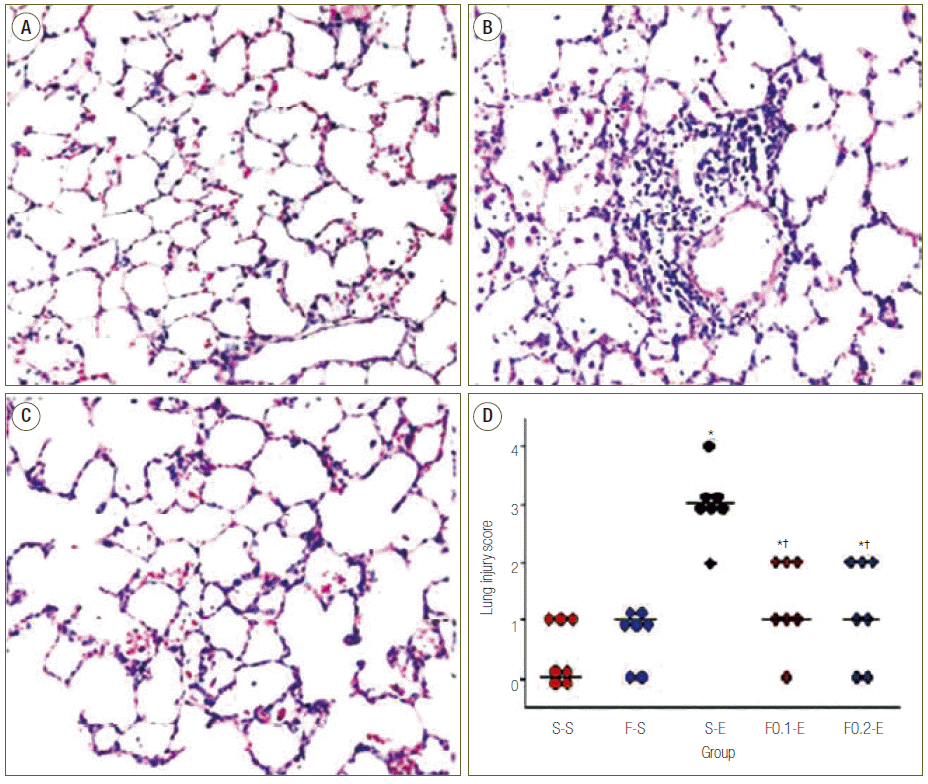
Endotoxin caused edema and hemorrhage of lung, thickening of the alveolar wall, and invasion of inflammatory cells into alveolar air spaces. However, flecainide prevented these changes (Fig. 2A-C). Histopathologic assessment of the LIS demonstrated that flecainide successfully attenuated the severity of the lung injury. (Table 1, Fig. 2D).
3) Analysis of BALF
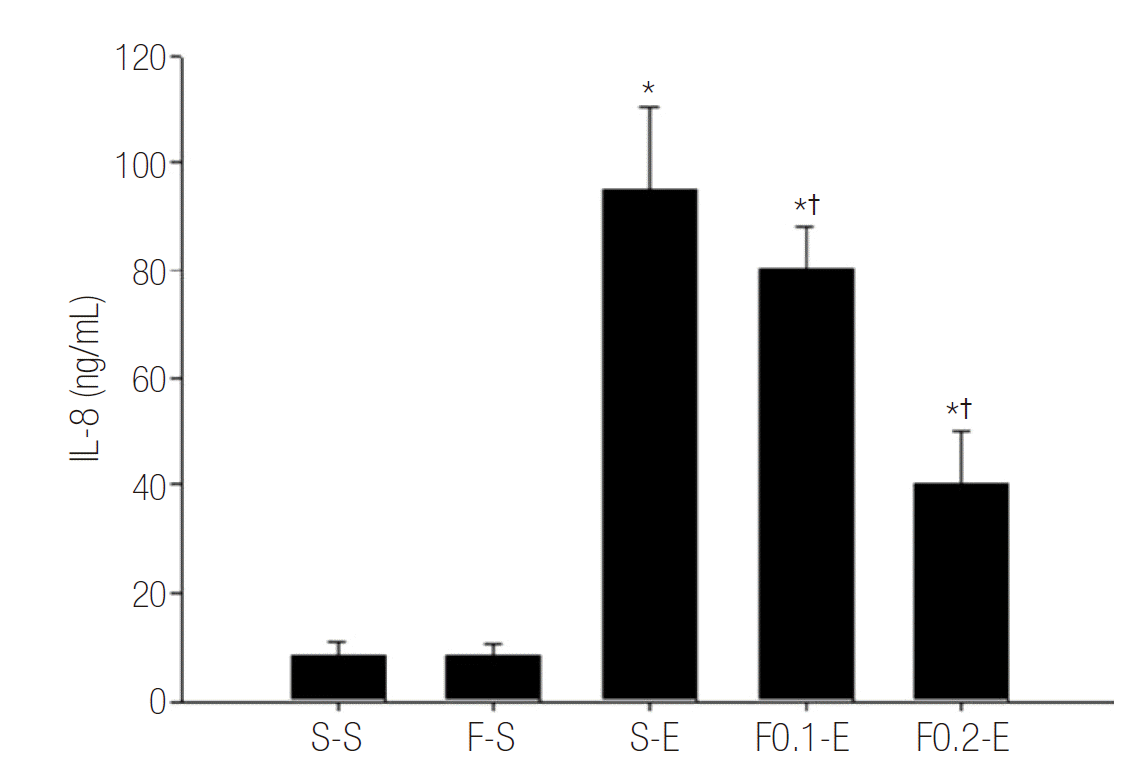
The ratio of BALFs collected from the animals was 77 to 85%. This range of ratio indicates a similar dilution among the groups. Obviously more leukocytes in BALF were collected in groups receiving endotoxin compared than in groups receiving saline, meaning that the neutrophils had invaded into the alveolar air spaces from the pulmonary capillary endothelium. And flecainide slightly prevented the increase in leukocyte counts in BALF (Table 1, Fig. 3A). Leukocytes of BALF in groups receiving saline were mostly macrophages. However, the ratio of PMNs in BALF markedly increased in groups receiving endotoxin in which the mean ratio of PMNs was as high as 41%. Flecainide significantly prevented the increase in these ratios (Table 1, Fig. 3B). The levels of IL-8 in BALF were increased in groups receiving endotoxin compared with in groups receiving saline. Flecainide prevented the increment in these concentrations (Table 1, Fig. 4).
Discussion
The main results in this experiment were that flecainide improved sepsis induced acute lung injury as analysed by W/D ratio and LIS, and inflammatory response as determined by leukocyte, PMNs and IL-8 in BALF. ARDS is a complex syndrome that characterize as severe pulmonary inflammatory responses with interstitial edema, damage of epithelial integrity, leukocyte infiltration and leakage of protein into alveolar air space and induce alterations in gas exchange.[9] Lipopolysaccharide (LPS) is induced in the host on infection with bacteria and it provokes inflammatory process which activate leukocyte, multiple kinases and proinflammatory cytokines involved with the expression of Toll-like receptors (TLR) 4 in alveolar epithelial cells.[10-12] The inflammatory event was a main cause of alterations in the processes which control lung fluid homeostasis. And eventually the alterations induce lung disease such as ARDS. In this experiment, flecainide decreased the degree of change of lung W/D ratio, histopathologic finding. Also it attenuated concentrations of leukocyte, PMNs and IL-8 in BALF. These findings are very important because these parameters are representative indexes of lung injury induced by disruption of cell integrity. Also in patients with sepsis and ALI, increased levels of leukocyte, PMNs and IL-8 correlate to a poor outcome.[13,14] Although a classically described role for flecainide is antiarrythmic action mediated by sodium channel blocker, we suggested that flecainide particularly appears to participate to regulate lung fluid homeostasis and neutrophil mediated inflammatory processes.
In other words, we proposed the hypothesis that the efficacy of flecainide was extend to decrease pulmonary edema and anti-inflammatory effects related with neutrophil activation. O’Brodovich[15] reported that LPS treated alveolar macrophages could attenuate total and amiloride sensitive sodium channel current in primary cultures of distal pulmonary epithelial cells. And Baines et al.[16] reported that LPS reduce epithelial sodium channel (ENaC) transcription and activity via extracellular signal regulated kinases (ERK) pathways. The alveolar ENaCs acts to resolve pulmonary edema via AFC.[2] Concept of AFC has emerged to explain lung fluid balance under both normal and pathologic conditions. Pathophysiologically, uncontrolled inflammation develop pulmonary edema and it provoke impairment of gas exchange and oxygenation in ALI and ARDS.[17] The failure of reabsorption of excess alveolar fluid to resolve this edema increase mortality in an intensive care unit.[18,19] In the concrete, through apical amiloride sensitive sodium channels, sodium ion enters the alveolar epithelial cells and it is actively transported across the basolateral membrane by the sodium-potassium-adenosine triphosphatase.[20] This vectorial transport of sodium is the main driving force for removal of edema from the air spaces. In some animal studies, it was reported that transgenic expression of ENaC mice (knock out mice) showed an exaggerated increase of the W/D ratio and more severe pulmonary edema on histological examination. These results are coincidence with our data for histopathologic finding that the pulmonary edema and hemorrhage of lung, thickening of the alveolar wall, and invasion of inflammatory cells were more prominent in rats receiving endotoxin. In view of above evidences about transepithelial Na+ transport, there are strong relations the ENaC with the pulmonary edema obviously. [21] Neutrophils, exposured to LPS, induce activation of multiple kinases which is involved in proinflammatory cytokine expression. Also oxygen radicals, protease, leukotrienes, and inflammatory cytokines, such as IL-1β, IL-8, TNF-α were released by activated neutrophils.[22] So, pathways inducing phosphorylation and activation of mitogen-activated protein kinases (MAPKs) such as p38, ERK 1/2 and c-Jun N-terminal kinase act to be critical to treat lung injury. Because LPS induced cytokine production by neutrophils is decreased by inhibition of activation of MAPKs.[23,24] A series of expression mediated with TLR-2 and 4 pathway are partially connected to the epithelial transport of sodium ion. In general, sodium ion shift in epithelial cell via regulation of several factors which is the regulation of the open probability, membrane expression and internalization of the ENaC and membrane expression and turnover of the sodium-potassium pump.[25] The sodium channel activity is not fully known in epithelium, but proinflammatory cytokine, serine protease, the thickness and osmolarity of the airway surface liquid contribute to regulate ENaC, partially. Especially, many of these factors, which are mediated with expression of TLR-2 and 4 pathway, including TNF-α,[26] IL-1,[27] IL-4, and nitric oxide, control the rate of airway epithelial sodium ion transport. Also activation of MAPK pathways, NF-κB and phosphorylation of ERK1/2 have been shown to modulate and interact the transcription and activity of ENaC in pulmonary epithelial cells.[28] Thus, In addition to its electrophysiologic effects, we propose that flecainide has major roles in attenuating of LPS induced neutrophil activation. However, the pathway and the specific roles of flecainide on inflammatory responses remain to be defined. So additional in vitro study should be performed to clarify these anti-inflammatory effects of flecainide acetate.
In conclusion, as showing in the present results, flecainide improve sepsis-induced lung injury in rats by controlling inflammatory response.
NOTES
-
No potential conflict of interest relevant to this article was reported.
Acknowledgments
This study was financially supported by Chonnam National University, 2014.
Fig. 1.Effects of flecainide acetate on endotoxin-induced changes in the wet-to-dry lung weight ratio (W/D ratio). Lung lobe was collected at 8 hour after the start of saline or endotoxin injection. W/D ratio was determined as described in Methods. Each values represent mean ± standard error of the mean from seven animals. *p < 0.05 vs. S-S group. †p < 0.05 vs. group S-E.
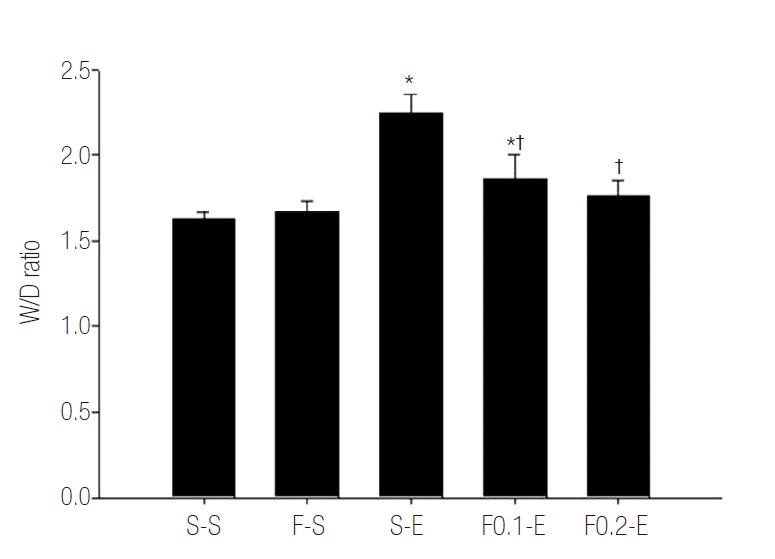
Fig. 2.Effects of flecainide acetate on lung tissue damage at 8 hour after the start of saline or endotoxin injection. Representive photomicrographies showing hematoxylin and eosin staining samples with median values in group S-S (A), group S-E (B), and group F-E (C). Original print magnifications x 400. (D) Lung injury score: lung injure was scored from 0 (no damage) to 4+ (maximal damage) according to the criteria described in Methods. Bars represent median from seven animals. *p < 0.05 vs. group S-S. †p < 0.05 vs. group S-E.
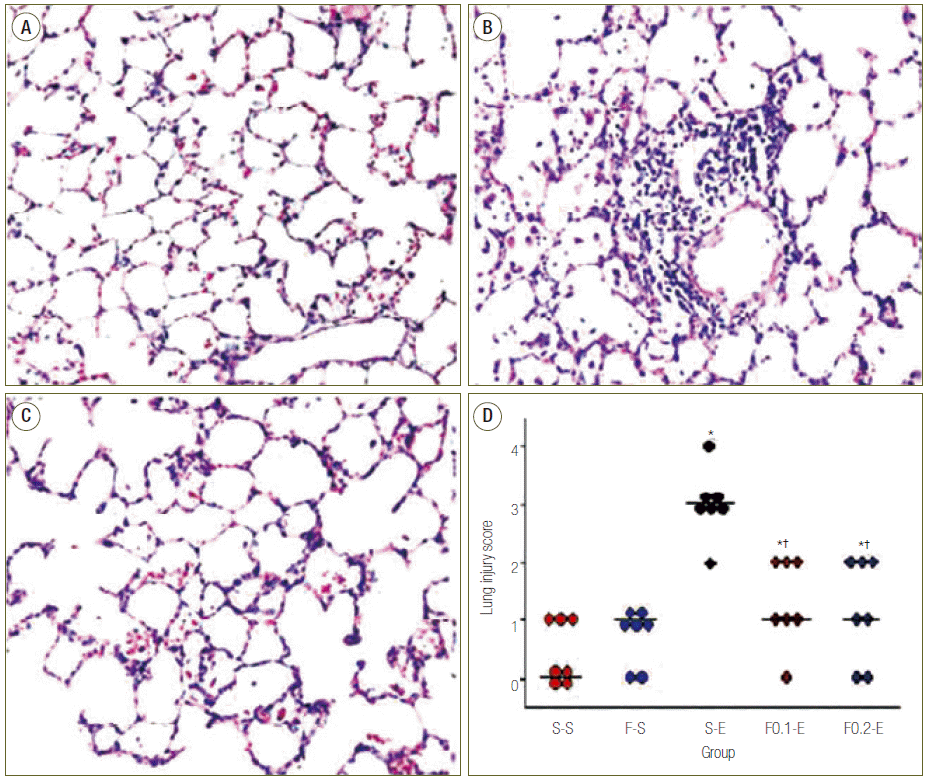
Fig. 3.Effects of flecainide acetate on endotoxin-induced changes in the leukocyte count (A) and percentage of polymorphonuclear neutrophils (PMNs) (B) in bronchoalveolar lavage fluids (BALF). BALF was collected at 8 hour after the start of saline or endotoxin injection. The leukocyte counts and percentage of PMNs in BALF were determined as described in Methods. The values represent mean ± standard error of the mean from seven animals. *p < 0.05 vs. S-S group. †p < 0.05 vs. group S-E.
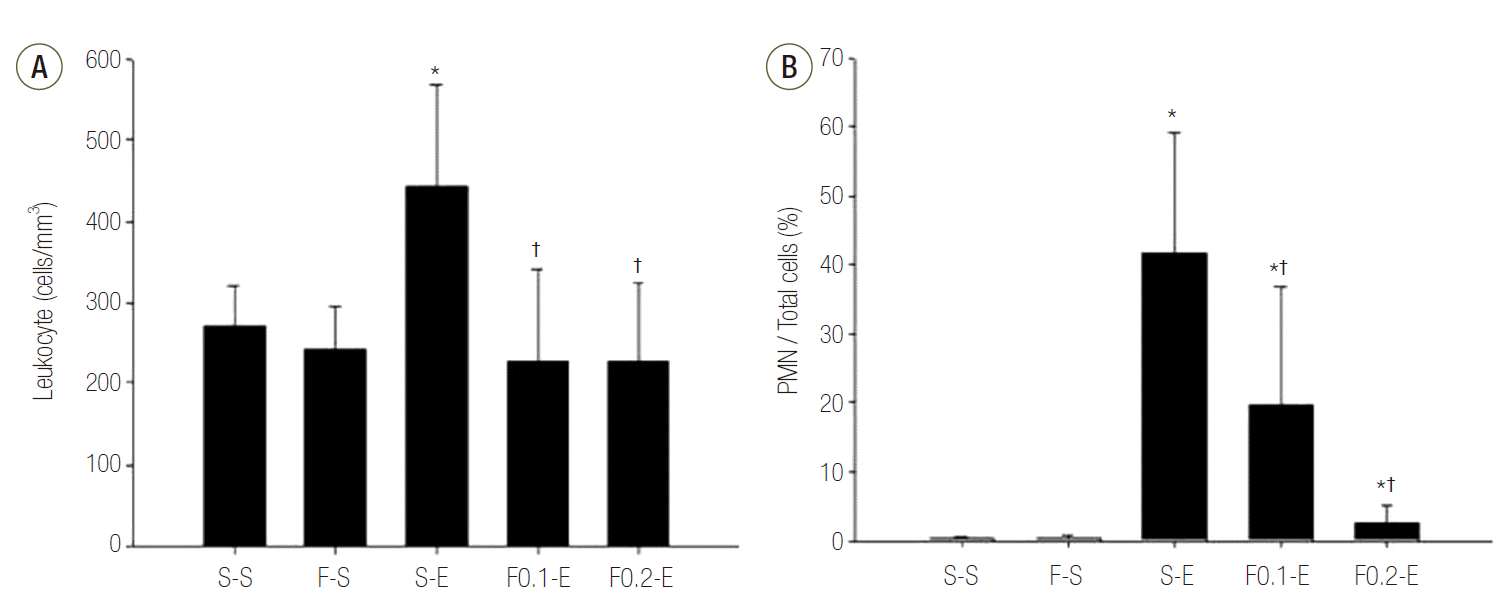
Fig. 4.Effects of flecainide acetate on endotoxin-induced changes in the concentration of Interleukin-8 (IL-8) in bronchoalveolar lavage fluids (BALF). BALF was collected at 8 hour after the start of saline or endotoxin injection. Concentration of IL-8 in BALF was determined as described in Methods. The values represent mean ± standard error of the mean from seven animals. *p < 0.05 vs. S-S group. †p < 0.05 vs. group S-E.
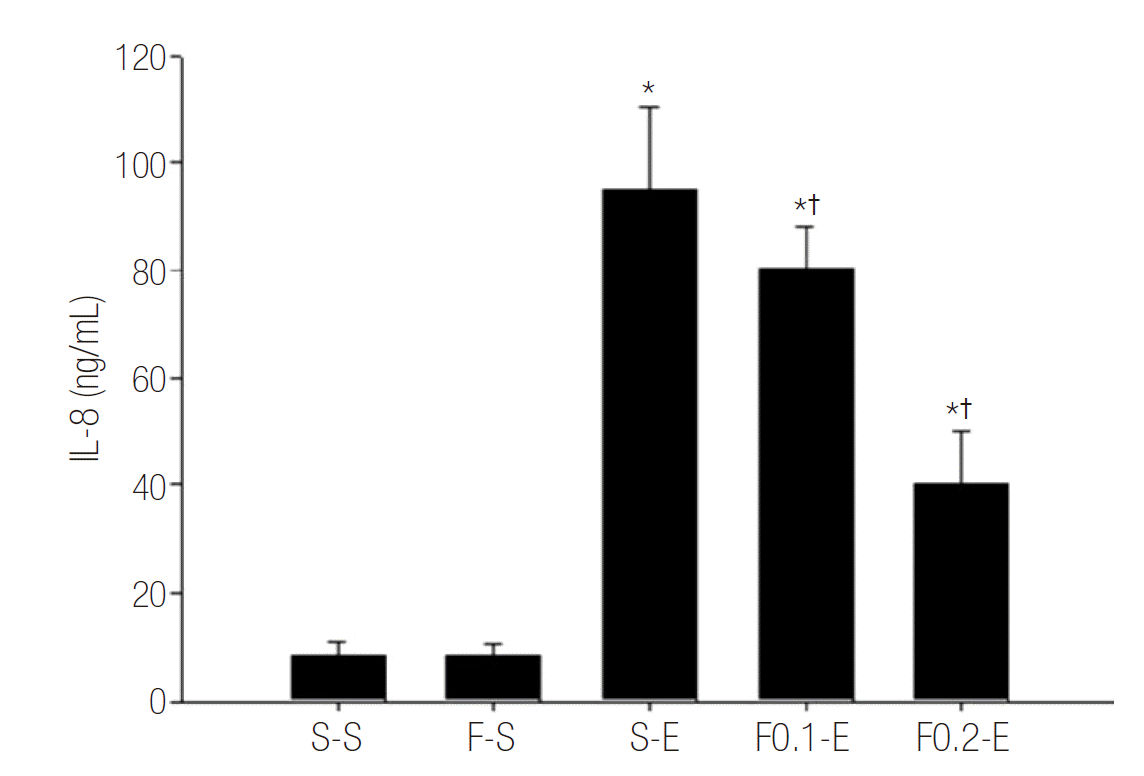
Table 1.Changes in BALF chemistry, W/D ratio, LIS in endotoxin and flecainide treated rats
|
Variables |
S-S |
F-S |
S-E |
F0.1-E |
F0.2-E |
|
BALF |
|
|
|
|
|
|
Leukocyte (×102/mm3) |
271 ± 49 |
243 ± 53 |
443 ± 127*
|
229 ± 111†
|
229 ± 95†
|
|
PMN/Total cells (%) |
0.39 ± 0.17 |
0.36 ± 0.34 |
41.43 ± 17.63*
|
19.5 ± 17.19*†
|
2.43 ± 2.61*†
|
|
IL-8 (ng/mL) |
8.60 ± 2.25 |
8.30 ± 2.20 |
95.00 ± 15.28*
|
80.37 ± 7.71*†
|
40.00 ± 10.21*†
|
|
W/D ratio |
1.63 ± 0.04 |
1.67 ± 0.06 |
2.24 ± 0.11*
|
1.86 ± 0.14*†
|
1.76 ± 0.09†
|
|
LIS (median [range]) |
0 (0-1) |
1 (0-1) |
3 (2-4)*
|
1 (0-2)*†
|
1 (0-2)*†
|
References
- 1. Held HD, Uhlig S. Mechanisms of endotoxin-induced airway and pulmonary vascular hyperreactivity in mice. Am J Respir Crit Care Med 2000;162(4 Pt 1):1547-52.ArticlePubMed
- 2. Kosari F, Sheng S, Li J, Mak DO, Foskett JK, Kleyman TR. Subunit stoichiometry of the epithelial sodium channel. J Biol Chem 1998;273:13469-74.ArticlePubMed
- 3. Samlowski WE, Frame RN, Logue GL. Flecainide-induced immune neutropenia. Documentation of a hapten-mediated mechanism of cell destruction. Arch Intern Med 1987;147:383-4.ArticlePubMed
- 4. Kwak SH, Mitra S, Bdeir K, Strassheim D, Park JS, Kim JY, et al. The kringle domain of urokinase-type plasminogen activator potentiates LPS-induced neutrophil activation through interaction with {alpha} V{beta}3 integrins. J Leukoc Biol 2005;78:937-45.ArticlePubMed
- 5. Abraham E, Arcaroli J, Carmody A, Wang H, Tracey KJ. HMG-1 as a mediator of acute lung inflammation. J Immunol 2000;165:2950-4.ArticlePubMed
- 6. Rowe SJ, Allen L, Ridger VC, Hellewell PG, Whyte MK. Caspase-1-deficient mice have delayed neutrophil apoptosis and a prolonged inflammatory response to lipopolysaccharide-induced acute lung injury. J Immunol 2002;169:6401-7.ArticlePubMed
- 7. Asti C, Ruggieri V, Porzio S, Chiusaroli R, Melillo G, Caselli GF. Lipopolysaccharide-induced lung injury in mice. I. Concomitant evaluation of inflammatory cells and haemorrhagic lung damage. Pulm Pharmacol Ther 2000;13:61-9.ArticlePubMed
- 8. Szarka RJ, Wang N, Gordon L, Nation PN, Smith RH. A murine model of pulmonary damage induced by lipopolysaccharide via intranasal instillation. J Immunol Methods 1997;202:49-57.ArticlePubMed
- 9. Kollef MH, Schuster DP. The acute respiratory distress syndrome. N Engl J Med 1995;332:27-37.ArticlePubMed
- 10. Saito T, Yamamoto T, Kazawa T, Gejyo H, Naito M. Expression of toll-like receptor 2 and 4 in lipopolysaccharide-induced lung injury in mouse. Cell Tissue Res 2005;321:75-88.ArticlePubMed
- 11. Guillot L, Medjane S, Le-Barillec K, Balloy V, Danel C, Chignard M, et al. Response of human pulmonary epithelial cells to lipopolysaccharide involves Toll-like receptor 4 (TLR4)-dependent signaling pathways: evidence for an intracellular compartmentalization of TLR4. J Biol Chem 2004;279:2712-8.ArticlePubMed
- 12. Roos-Engstrand E, Wallin A, Bucht A, Pourazar J, Sandström T, Blomberg A. Increased expression of p38 MAPK in human bronchial epithelium after lipopolysaccharide exposure. Eur Respir J 2005;25:797-803.ArticlePubMed
- 13. Donnelly SC, Strieter RM, Kunkel SL, Walz A, Robertson CR, Carter DC, et al. Interleukin-8 and development of adult respiratory distress syndrome in atrisk patient groups. Lancet 1993;341:643-7.ArticlePubMed
- 14. Reinhart K, Meisner M, Brunkhorst FM. Markers for sepsis diagnosis: what is useful? Crit Care Clin 2006;22:503. -19. ix-x.ArticlePubMed
- 15. O’Brodovich HM. The role of active Na+ transport by lung epithelium in the clearance of airspace fluid. New Horiz 1995;3:240-7.PubMed
- 16. Baines DL, Albert AP, Hazell MJ, Gambling L, Woollhead AM, Dockrell ME. Lipopolysaccharide modifies amiloride-sensitive Na+ transport processes across human airway cells: role of mitogen-activated protein kinases ERK 1/2 and 5. Pflugers Arch 2010;459:451-63.ArticlePubMedPMC
- 17. Wyncoll DL, Evans TW. Acute respiratory distress syndrome. Lancet 1999;354:497-501.ArticlePubMed
- 18. Ware LB, Matthay MA. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med 2001;163:1376-83.ArticlePubMed
- 19. Sznajder JI. Alveolar edema must be cleared for the acute respiratory distress syndrome patient to survive. Am J Respir Crit Care Med 2001;163:1293-4.ArticlePubMed
- 20. Matthay MA, Clerici C, Saumon G. Invited review: Active fluid clearance from the distal air spaces of the lung. J Appl Physiol 2002;93:1533-41.ArticlePubMed
- 21. Egli M, Duplain H, Lepori M, Cook S, Nicod P, Hummler E, et al. Defective respiratory amiloride-sensitive sodium transport predisposes to pulmonary oedema and delays its resolution in mice. J Physiol 2004;560(Pt 3):857-65.ArticlePubMedPMC
- 22. Abraham E, Carmody A, Shenkar R, Arcaroli J. Neutrophils as early immunologic effectors in hemorrhage- or endotoxemia-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol 2000;279:L1137-45.ArticlePubMed
- 23. Yum HK, Arcaroli J, Kupfner J, Shenkar R, Penninger JM, Sasaki T, et al. Involvement of phosphoinositide 3-kinases in neutrophil activation and the development of acute lung injury. J Immunol 2001;167:6601-8.ArticlePubMed
- 24. Selvatici R, Falzarano S, Mollica A, Spisani S. Signal transduction pathways triggered by selective formylpeptide analogues in human neutrophils. Eur J Pharmacol 2006;534:1-11.ArticlePubMed
- 25. Geering K. Functional roles of Na,K-ATPase subunits. Curr Opin Nephrol Hypertens 2008;17:526-32.ArticlePubMed
- 26. Dagenais A, Fréchette R, Yamagata Y, Yamagata T, Carmel JF, Clermont ME, et al. Downregulation of ENaC activity and expression by TNF-alpha in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 2004;286:L301-11.ArticlePubMed
- 27. Ye X, Acharya R, Herbert JB, Hamilton SE, Folkesson HG. IL-1beta stimulates alveolar fluid absorption in fetal guinea pig lungs via the hypothalamuspituitary-adrenal gland axis. Am J Physiol Lung Cell Mol Physiol 2004;286:L756-66.ArticlePubMed
- 28. de Seigneux S, Leroy V, Ghzili H, Rousselot M, Nielsen S, Rossier BC, et al. NF-kappaB inhibits sodium transport via down-regulation of SGK1 in renal collecting duct principal cells. J Biol Chem 2008;283:25671-81.ArticlePubMed
Citations
Citations to this article as recorded by





 PubReader
PubReader ePub Link
ePub Link Cite
Cite