Articles
- Page Path
- HOME > Acute Crit Care > Volume 37(1); 2022 > Article
-
Review Article
Neurosurgery Brain-lung interaction: a vicious cycle in traumatic brain injury -
Ariana Alejandra Chacón-Aponte1
 , Érika Andrea Durán-Vargas1, Jaime Adolfo Arévalo-Carrillo1, Iván David Lozada-Martínez2,3,4,5, Maria Paz Bolaño-Romero5, Luis Rafael Moscote-Salazar2,3,5, Pedro Grille6, Tariq Janjua7
, Érika Andrea Durán-Vargas1, Jaime Adolfo Arévalo-Carrillo1, Iván David Lozada-Martínez2,3,4,5, Maria Paz Bolaño-Romero5, Luis Rafael Moscote-Salazar2,3,5, Pedro Grille6, Tariq Janjua7 -
Acute and Critical Care 2022;37(1):35-44.
DOI: https://doi.org/10.4266/acc.2021.01193
Published online: February 11, 2022
1Colombian Clinical Research Group in Neurocritical Care, University of Pamplona, Cúcuta, Colombia
2Colombian Clinical Research Group in Neurocritical Care, University of Cartagena, Cartagena, Colombia
3Latin American Council of Neurocritical Care (CLaNi), Cartagena, Colombia
4Global Neurosurgery Committee, World Federation of Neurosurgical Societies, Cartagena, Colombia
5Medical and Surgical Research Center, Cartagena, Colombia
6Department of Intensive Care, Hospital Maciel, Montevideo, Uruguay
7Department of Intensive Care, Regions Hospital, St. Paul, MN, USA
- Corresponding author: Iván David Lozada-Martínez Colombian Clinical Research Group in Neurocritical Care, University of Cartagena, Cra. 50 #24120, Cartagena 130004, Colombia Tel: +57-3157799823, E-mail: ilozadam@unicartagena.edu.co
Copyright © 2022 The Korean Society of Critical Care Medicine
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
- The brain-lung interaction can seriously affect patients with traumatic brain injury, triggering a vicious cycle that worsens patient prognosis. Although the mechanisms of the interaction are not fully elucidated, several hypotheses, notably the “blast injury” theory or “double hit” model, have been proposed and constitute the basis of its development and progression. The brain and lungs strongly interact via complex pathways from the brain to the lungs but also from the lungs to the brain. The main pulmonary disorders that occur after brain injuries are neurogenic pulmonary edema, acute respiratory distress syndrome, and ventilator-associated pneumonia, and the principal brain disorders after lung injuries include brain hypoxia and intracranial hypertension. All of these conditions are key considerations for management therapies after traumatic brain injury and need exceptional case-by-case monitoring to avoid neurological or pulmonary complications. This review aims to describe the history, pathophysiology, risk factors, characteristics, and complications of brain-lung and lung-brain interactions and the impact of different old and recent modalities of treatment in the context of traumatic brain injury.
INTRODUCTION
HISTORY
BRAIN-LUNG INTERACTIONS IN BRAIN TRAUMA
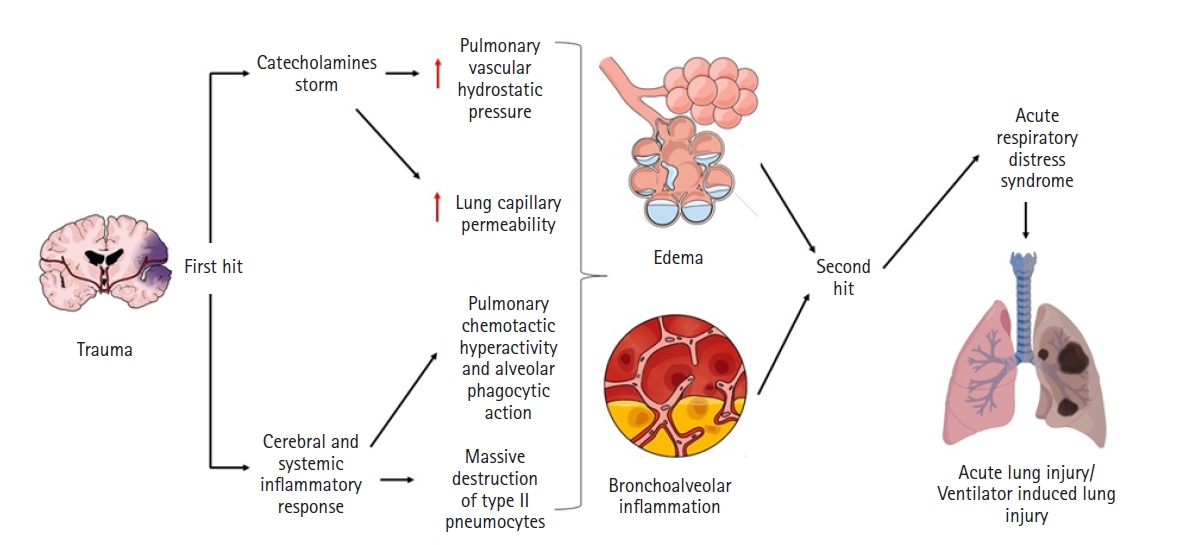
Physiopathology
Neurogenic pulmonary edema
Acute respiratory distress
Ventilation-associated pneumonia
Physiopathology
Cerebral hypoxia
Intracranial hypertension
Therapeutic Implications: the Conflict between the Brain and Lungs
CONCLUSION
KEY MESSAGES
-
CONFLICT OF INTEREST
No potential conflict of interest relevant to this article was reported.
-
AUTHOR CONTRIBUTIONS
Conceptualization: all authors. Data curation: AACA, EADV, JAAC, IDLM, MPBR. Formal analysis: AACA, EADV, JAAC, IDLM, MPBR. Methodology: all authors. Project administration: IDLM, LRMS, PG, TJ. Writing–original draft: all authors. Writing–review & editing: all authors.
NOTES
- 1. Johnson WD, Griswold DP. Traumatic brain injury: a global challenge. Lancet Neurol 2017;16:949-50.ArticlePubMed
- 2. Okidi R, Ogwang DM, Okello TR, Ezati D, Kyegombe W, Nyeko D, et al. Factors affecting mortality after traumatic brain injury in a resource-poor setting. BJS Open 2020;4:320-5.ArticlePubMed
- 3. Lee K, Rincon F. Pulmonary complications in patients with severe brain injury. Crit Care Res Pract 2012;2012:207247. ArticlePubMedPMC
- 4. Mrozek S, Constantin JM, Geeraerts T. Brain-lung crosstalk: implications for neurocritical care patients. World J Crit Care Med 2015;4:163-78.ArticlePubMedPMC
- 5. Brown-Séquard CE. On the production of hemorrhage, anemia, edema and emphysema in the lungs by injuries to the base of the brain. Lancet 1871;97:6. Article
- 6. Simmons RL, Martin AM Jr, Heisterkamp CA 3rd, Ducker TB. Respiratory insufficiency in combat casualties. II. Pulmonary edema following head injury. Ann Surg 1969;170:39-44.PubMedPMC
- 7. Theodore J, Robin ED. Speculations on neurogenic pulmonary edema (NPE). Am Rev Respir Dis 1976;113:405-11.PubMed
- 8. Brambrink AM, Dick WF. Neurogenic pulmonary edema: pathogenesis, clinical picture and therapy. Anaesthesist 1997;46:953-63.PubMed
- 9. López-Aguilar J, Fernández-Gonzalo MS, Turon M, Quílez ME, Gómez-Simón V, Jódar MM, et al. Lung-brain interaction in the mechanically ventilated patient. Med Intensiva 2013;37:485-92.ArticlePubMed
- 10. Ashbaugh DG, Bigelow DB, Petty TL, Levine BE. Acute respiratory distress in adults. Lancet 1967;2:319-23.ArticlePubMed
- 11. Murray JF, Matthay MA, Luce JM, Flick MR. An expanded definition of the adult respiratory distress syndrome. Am Rev Respir Dis 1988;138:720-3.ArticlePubMed
- 12. Bratton SL, Davis RL. Acute lung injury in isolated traumatic brain injury. Neurosurgery 1997;40:707-12.ArticlePubMed
- 13. Holland MC, Mackersie RC, Morabito D, Campbell AR, Kivett VA, Patel R, et al. The development of acute lung injury is associated with worse neurologic outcome in patients with severe traumatic brain injury. J Trauma 2003;55:106-11.ArticlePubMed
- 14. Contant CF, Valadka AB, Gopinath SP, Hannay HJ, Robertson CS. Adult respiratory distress syndrome: a complication of induced hypertension after severe head injury. J Neurosurg 2001;95:560-8.ArticlePubMed
- 15. Rincon F, Ghosh S, Dey S, Maltenfort M, Vibbert M, Urtecho J, et al. Impact of acute lung injury and acute respiratory distress syndrome after traumatic brain injury in the United States. Neurosurgery 2012;71:795-803.ArticlePubMed
- 16. Dziedzic T, Slowik A, Szczudlik A. Nosocomial infections and immunity: lesson from brain-injured patients. Crit Care 2004;8:266-70.ArticlePubMedPMC
- 17. Piek J, Chesnut RM, Marshall LF, van Berkum-Clark M, Klauber MR, Blunt BA, et al. Extracranial complications of severe head injury. J Neurosurg 1992;77:901-7.ArticlePubMed
- 18. Woratyla SP, Morgan AS, Mackay L, Bernstein B, Barba C. Factors associated with early onset pneumonia in the severely brain-injured patient. Conn Med 1995;59:643-7.PubMed
- 19. Fàbregas N, Torres A. Pulmonary infection in the brain injured patient. Minerva Anestesiol 2002;68:285-90.PubMed
- 20. Ranieri VM, Suter PM, Tortorella C, De Tullio R, Dayer JM, Brienza A, et al. Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome: a randomized controlled trial. JAMA 1999;282:54-61.ArticlePubMed
- 21. Bronchard R, Albaladejo P, Brezac G, Geffroy A, Seince PF, Morris W, et al. Early onset pneumonia: risk factors and consequences in head trauma patients. Anesthesiology 2004;100:234-9.PubMed
- 22. Koutsoukou A, Katsiari M, Orfanos SE, Kotanidou A, Daganou M, Kyriakopoulou M, et al. Respiratory mechanics in brain injury: a review. World J Crit Care Med 2016;5:65-73.ArticlePubMedPMC
- 23. Gonzalvo R, Martí-Sistac O, Blanch L, López-Aguilar J. Bench-to-bedside review: brain-lung interaction in the critically ill. A pending issue revisited. Crit Care 2007;11:216. ArticlePubMedPMC
- 24. Mascia L. Acute lung injury in patients with severe brain injury: a double hit model. Neurocrit Care 2009;11:417-26.ArticlePubMed
- 25. Al-Dhahir MA, Das JM, Sharma S. Neurogenic pulmonary edema [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 [cited 2021 Sep 31]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK532984/.
- 26. Kennedy JD, Hardin KA, Parikh P, Li CS, Seyal M. Pulmonary edema following generalized tonic clonic seizures is directly associated with seizure duration. Seizure 2015;27:19-24.ArticlePubMedPMC
- 27. Raja HM, Herwadkar AV, Paroutoglou K, Lilleker JB. Neurogenic pulmonary oedema complicating a lateral medullary infarct. BMJ Case Rep 2018;2018:bcr2018225437. ArticlePubMedPMC
- 28. Romero Osorio OM, Abaunza Camacho JF, Sandoval Briceño D, Lasalvia P, Narino Gonzalez D. Postictal neurogenic pulmonary edema: case report and brief literature review. Epilepsy Behav Case Rep 2017;9:49-50.ArticlePubMedPMC
- 29. Šedý J, Kuneš J, Zicha J. Pathogenetic mechanisms of neurogenic pulmonary edema. J Neurotrauma 2015;32:1135-45.ArticlePubMed
- 30. Busl KM, Bleck TP. Neurogenic pulmonary edema. Crit Care Med 2015;43:1710-5.ArticlePubMed
- 31. Zhang L, Yao J, Zhang T, Jin J, Zeng X, Yue Z. Stellate ganglion block may prevent the development of neurogenic pulmonary edema and improve the outcome. Med Hypotheses 2013;80:158-61.ArticlePubMed
- 32. Rincon F, Maltenfort M, Dey S, Ghosh S, Vibbert M, Urtecho J, et al. The prevalence and impact of mortality of the acute respiratory distress syndrome on admissions of patients with ischemic stroke in the United States. J Intensive Care Med 2014;29:357-64.ArticlePubMed
- 33. Papazian L, Klompas M, Luyt CE. Ventilator-associated pneumonia in adults: a narrative review. Intensive Care Med 2020;46:888-906.ArticlePubMedPMC
- 34. Othman AA, Abdelazim MS. Ventilator-associated pneumonia in adult intensive care unit prevalence and complications. Egypt J Crit Care Med 2017;5:61-3.Article
- 35. Ding C, Zhang Y, Yang Z, Wang J, Jin A, Wang W, et al. Incidence, temporal trend and factors associated with ventilator-associated pneumonia in mainland China: a systematic review and meta-analysis. BMC Infect Dis 2017;17:468. ArticlePubMedPMC
- 36. Jovanovic B, Milan Z, Markovic-Denic L, Djuric O, Radinovic K, Doklestic K, et al. Risk factors for ventilator-associated pneumonia in patients with severe traumatic brain injury in a Serbian trauma centre. Int J Infect Dis 2015;38:46-51.ArticlePubMed
- 37. Di Pasquale M, Ferrer M, Esperatti M, Crisafulli E, Giunta V, Li Bassi G, et al. Assessment of severity of ICU-acquired pneumonia and association with etiology. Crit Care Med 2014;42:303-12.ArticlePubMed
- 38. Huang Y, Jiao Y, Zhang J, Xu J, Cheng Q, Li Y, et al. Microbial etiology and prognostic factors of ventilator-associated pneumonia: a multicenter retrospective study in Shanghai. Clin Infect Dis 2018;67(Suppl 2):S146-52.ArticlePubMed
- 39. Kalil AC, Metersky ML, Klompas M, Muscedere J, Sweeney DA, Palmer LB, et al. Management of adults with hospital-acquired and ventilator-associated pneumonia: 2016 clinical practice guidelines by the Infectious Diseases Society of America and the American Thoracic Society. Clin Infect Dis 2016;63:e61-111.ArticlePubMedPMCPDF
- 40. Martin-Loeches I, Deja M, Koulenti D, Dimopoulos G, Marsh B, Torres A, et al. Potentially resistant microorganisms in intubated patients with hospital-acquired pneumonia: the interaction of ecology, shock and risk factors. Intensive Care Med 2013;39:672-81.ArticlePubMed
- 41. Bickenbach J, Zoremba N, Fries M, Dembinski R, Doering R, Ogawa E, et al. Low tidal volume ventilation in a porcine model of acute lung injury improves cerebral tissue oxygenation. Anesth Analg 2009;109:847-55.ArticlePubMed
- 42. Mikkelsen ME, Christie JD, Lanken PN, Biester RC, Thompson BT, Bellamy SL, et al. The adult respiratory distress syndrome cognitive outcomes study: long-term neuropsychological function in survivors of acute lung injury. Am J Respir Crit Care Med 2012;185:1307-15.ArticlePubMedPMC
- 43. Fries M, Bickenbach J, Henzler D, Beckers S, Dembinski R, Sellhaus B, et al. S-100 protein and neurohistopathologic changes in a porcine model of acute lung injury. Anesthesiology 2005;102:761-7.ArticlePubMed
- 44. Bickenbach J, Biener I, Czaplik M, Nolte K, Dembinski R, Marx G, et al. Neurological outcome after experimental lung injury. Respir Physiol Neurobiol 2011;179:174-80.ArticlePubMed
- 45. Bassi TG, Rohrs EC, Reynolds SC. Systematic review of cognitive impairment and brain insult after mechanical ventilation. Crit Care 2021;25:99. ArticlePubMedPMC
- 46. Hopkins RO, Gale SD, Weaver LK. Brain atrophy and cognitive impairment in survivors of acute respiratory distress syndrome. Brain Inj 2006;20:263-71.ArticlePubMed
- 47. Kamuf J, Garcia-Bardon A, Ziebart A, Thomas R, Folkert K, Frauenknecht K, et al. Lung injury does not aggravate mechanical ventilation-induced early cerebral inflammation or apoptosis in an animal model. PLoS One 2018;13:e0202131. ArticlePubMedPMC
- 48. Al-Attar R, Childers CL, Nguyen VC, Pamenter ME, Storey KB. Differential protein phosphorylation is responsible for hypoxia-induced regulation of the Akt/mTOR pathway in naked mole rats. Comp Biochem Physiol A Mol Integr Physiol 2020;242:110653. ArticlePubMed
- 49. Caricato A, Conti G, Della Corte F, Mancino A, Santilli F, Sandroni C, et al. Effects of PEEP on the intracranial system of patients with head injury and subarachnoid hemorrhage: the role of respiratory system compliance. J Trauma 2005;58:571-6.ArticlePubMed
- 50. Heuer JF, Pelosi P, Hermann P, Perske C, Crozier TA, Brück W, et al. Acute effects of intracranial hypertension and ARDS on pulmonary and neuronal damage: a randomized experimental study in pigs. Intensive Care Med 2011;37:1182-91.ArticlePubMedPMC
- 51. Robba C, Poole D, McNett M, Asehnoune K, Bösel J, Bruder N, et al. Mechanical ventilation in patients with acute brain injury: recommendations of the European Society of Intensive Care Medicine consensus. Intensive Care Med 2020;46:2397-410.ArticlePubMedPMC
- 52. Della Torre V, Badenes R, Corradi F, Racca F, Lavinio A, Matta B, et al. Acute respiratory distress syndrome in traumatic brain injury: how do we manage it? J Thorac Dis 2017;9:5368-81.ArticlePubMedPMC
- 53. Mascia L, Zavala E, Bosma K, Pasero D, Decaroli D, Andrews P, et al. High tidal volume is associated with the development of acute lung injury after severe brain injury: an international observational study. Crit Care Med 2007;35:1815-20.ArticlePubMed
- 54. Krebs J, Tsagogiorgas C, Pelosi P, Rocco PR, Hottenrott M, Sticht C, et al. Open lung approach with low tidal volume mechanical ventilation attenuates lung injury in rats with massive brain damage. Crit Care 2014;18:R59. ArticlePubMedPMC
- 55. Borsellino B, Schultz MJ, Gama de Abreu M, Robba C, Bilotta F. Mechanical ventilation in neurocritical care patients: a systematic literature review. Expert Rev Respir Med 2016;10:1123-32.ArticlePubMed
- 56. Carney N, Totten AM, O'Reilly C, Ullman JS, Hawryluk GW, Bell MJ, et al. Guidelines for the management of severe traumatic brain injury, fourth edition. Neurosurgery 2017;80:6-15.ArticlePubMed
- 57. Asehnoune K, Roquilly A, Cinotti R. Respiratory management in patients with severe brain injury. Crit Care 2018;22:76. ArticlePubMedPMC
- 58. Chapin JC, Downs JB, Douglas ME, Murphy EJ, Ruiz BC. Lung expansion, airway pressure transmission, and positive end-expiratory pressure. Arch Surg 1979;114:1193-7.ArticlePubMed
- 59. Nemer SN, Caldeira JB, Santos RG, Guimarães BL, Garcia JM, Prado D, et al. Effects of positive end-expiratory pressure on brain tissue oxygen pressure of severe traumatic brain injury patients with acute respiratory distress syndrome: A pilot study. J Crit Care 2015;30:1263-6.ArticlePubMed
- 60. Chen H, Chen K, Xu JQ, Zhang YR, Yu RG, Zhou JX. Intracranial pressure responsiveness to positive end-expiratory pressure is influenced by chest wall elastance: a physiological study in patients with aneurysmal subarachnoid hemorrhage. BMC Neurol 2018;18:124. ArticlePubMedPMC
- 61. Ashton-Cleary DT, Duffy MR. Prone ventilation for refractory hypoxaemia in a patient with severe chest wall disruption and traumatic brain injury. Br J Anaesth 2011;107:1009-10.ArticlePubMed
- 62. Robba C, Rebora P, Banzato E, Wiegers E, Stocchetti N, Menon DK, et al. Incidence, risk factors, and effects on outcome of ventilator-associated pneumonia in patients with traumatic brain injury: analysis of a large, multicenter, prospective, observational longitudinal study. Chest 2020;158:2292-303.PubMed
- 63. Kerr N, de Rivero Vaccari JP, Dietrich WD, Keane RW. Neural-respiratory inflammasome axis in traumatic brain injury. Exp Neurol 2020;323:113080. ArticlePubMed
- 64. Bortolotti P, Faure E, Kipnis E. Inflammasomes in tissue damages and immune disorders after trauma. Front Immunol 2018;9:1900. ArticlePubMedPMC
- 65. Lee SW, de Rivero Vaccari JP, Truettner JS, Dietrich WD, Keane RW. The role of microglial inflammasome activation in pyroptotic cell death following penetrating traumatic brain injury. J Neuroinflammation 2019;16:27. ArticlePubMedPMC
- 66. Kerr NA, de Rivero Vaccari JP, Weaver C, Dietrich WD, Ahmed T, Keane RW. Enoxaparin attenuates acute lung injury and inflammasome activation after traumatic brain injury. J Neurotrauma 2021;38:646-54.ArticlePubMedPMC
References
Figure & Data
References
Citations
- Uncertainty in Neurocritical Care: Recognizing Its Relevance for Clinical Decision-Making
Luis Rafael Moscote-Salazar, William A. Florez-Perdomo, Tariq Janjua
Indian Journal of Neurotrauma.2024; 21(01): 092. CrossRef - Manejo postoperatorio de resección de tumores cerebrales en la unidad de cuidado intensivo
Andrés Felipe Naranjo Ramírez, Álvaro de Jesús Medrano Areiza, Bryan Arango Sánchez, Juan Carlos Arango Martínez, Luis Fermín Naranjo Atehortúa
Acta Colombiana de Cuidado Intensivo.2024; 24(2): 140. CrossRef - Effects of positive end-expiratory pressure on intracranial pressure, cerebral perfusion pressure, and brain oxygenation in acute brain injury: Friend or foe? A scoping review
Greta Zunino, Denise Battaglini, Daniel Agustin Godoy
Journal of Intensive Medicine.2024; 4(2): 247. CrossRef - Acute brain injury increases pulmonary capillary permeability via sympathetic activation-mediated high fluid shear stress and destruction of the endothelial glycocalyx layer
Na Zhao, Chao Liu, Xinxin Tian, Juan Yang, Tianen Wang
Experimental Cell Research.2024; 434(2): 113873. CrossRef - Oral administration of lysozyme protects against injury of ileum via modulating gut microbiota dysbiosis after severe traumatic brain injury
Weijian Yang, Caihua Xi, Haijun Yao, Qiang Yuan, Jun Zhang, Qifang Chen, Gang Wu, Jin Hu
Frontiers in Cellular and Infection Microbiology.2024;[Epub] CrossRef - Pulmonary Effects of Traumatic Brain Injury in Mice: A Gene Set Enrichment Analysis
Wei-Hung Chan, Shih-Ming Huang, Yi-Lin Chiu
International Journal of Molecular Sciences.2024; 25(5): 3018. CrossRef - Beyond the brain: General intensive care considerations in pediatric neurocritical care
Thao L. Nguyen, Dennis W. Simon, Yi-Chen Lai
Seminars in Pediatric Neurology.2024; 49: 101120. CrossRef - Research Progress of Hemorrhagic Stroke Combined with Stroke-Associated Pneumonia
松 刘
Advances in Clinical Medicine.2024; 14(05): 2336. CrossRef - The Impact of Pulmonary Disorders on Neurological Health (Lung-Brain Axis)
Hongryeol Park, Chan Hee Lee
Immune Network.2024;[Epub] CrossRef - Modeling of the brain-lung axis using organoids in traumatic brain injury: an updated review
Jong-Tae Kim, Kang Song, Sung Woo Han, Dong Hyuk Youn, Harry Jung, Keun-Suh Kim, Hyo-Jung Lee, Ji Young Hong, Yong-Jun Cho, Sung-Min Kang, Jin Pyeong Jeon
Cell & Bioscience.2024;[Epub] CrossRef - Ventilatory targets following brain injury
Shaurya Taran, Sarah Wahlster, Chiara Robba
Current Opinion in Critical Care.2023; 29(2): 41. CrossRef - Targeted Nanocarriers Co-Opting Pulmonary Intravascular Leukocytes for Drug Delivery to the Injured Brain
Jia Nong, Patrick M. Glassman, Jacob W. Myerson, Viviana Zuluaga-Ramirez, Alba Rodriguez-Garcia, Alvin Mukalel, Serena Omo-Lamai, Landis R. Walsh, Marco E. Zamora, Xijing Gong, Zhicheng Wang, Kartik Bhamidipati, Raisa Y. Kiseleva, Carlos H. Villa, Colin F
ACS Nano.2023; 17(14): 13121. CrossRef - Modulation of MAPK/NF-κB Pathway and NLRP3 Inflammasome by Secondary Metabolites from Red Algae: A Mechanistic Study
Asmaa Nabil-Adam, Mohamed L. Ashour, Mohamed Attia Shreadah
ACS Omega.2023; 8(41): 37971. CrossRef - American Association for the Surgery of Trauma/American College of Surgeons Committee on Trauma clinical protocol for management of acute respiratory distress syndrome and severe hypoxemia
Jason A. Fawley, Christopher J. Tignanelli, Nicole L. Werner, George Kasotakis, Samuel P. Mandell, Nina E. Glass, David J. Dries, Todd W. Costantini, Lena M. Napolitano
Journal of Trauma and Acute Care Surgery.2023; 95(4): 592. CrossRef - The role of cardiac dysfunction and post-traumatic pulmonary embolism in brain-lung interactions following traumatic brain injury
Mabrouk Bahloul, Karama Bouchaala, Najeh Baccouche, Kamilia Chtara, Hedi Chelly, Mounir Bouaziz
Acute and Critical Care.2022; 37(2): 266. CrossRef - Allocation of Donor Lungs in Korea
Hye Ju Yeo
Journal of Chest Surgery.2022; 55(4): 274. CrossRef - Mapping brain endophenotypes associated with idiopathic pulmonary fibrosis genetic risk
Ali-Reza Mohammadi-Nejad, Richard J. Allen, Luke M. Kraven, Olivia C. Leavy, R. Gisli Jenkins, Louise V. Wain, Dorothee P. Auer, Stamatios N. Sotiropoulos
eBioMedicine.2022; 86: 104356. CrossRef - Use of bedside ultrasound in the evaluation of acute dyspnea: a comprehensive review of evidence on diagnostic usefulness
Ivan David Lozada-Martinez, Isabela Zenilma Daza-Patiño, Gerardo Jesus Farley Reina-González, Sebastián Rojas-Pava, Ailyn Zenith Angulo-Lara, María Paola Carmona-Rodiño, Olga Gissela Sarmiento-Najar, Jhon Mike Romero-Madera, Yesid Alonso Ángel-Hernandez
Revista Investigación en Salud Universidad de Boyacá.2022;[Epub] CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite