Abstract
-
Background
- Flecainide acetate is a drug used primarily for cardiac arrhythmia. Some studies also imply that flecainide acetate has the potential to regulate inflammatory-immune responses; however, its mechanism of action is contended. We determined the effects of flecainide acetate on lipopolysaccharide (LPS)-stimulated human neutrophils in vitro and on mortality in a septic rat model.
-
Methods
- Neutrophils from human blood were cultured with varying concentrations of flecainide acetate (1 μM, 10 μM, or 100 μM) with or without LPS (100 ng/ml). To assess neutrophil activation, the protein levels of tumor necrosis factor-alpha (TNF-α) and interleukin (IL)-6 and IL-8 were measured after a 4-hour culture period. To assess the intracellular signaling pathways, the levels of phosphorylation of p38 mitogen-activated protein kinase (p38), extracellular signal-regulated kinase (ERK) 1/2, and c-Jun N-terminal kinase (JNK) were measured after a 30-minute culture period, and the nuclear translocation of nuclear factor (NF)-κB was measured after a 1-hour culture period. Additionally, the survival rate was investigated in a rat sepsis model.
-
Results
- Flecainide acetate down-regulated the activation of proinflammatory cytokines, including TNF-α and IL-6 and IL-8, and intracellular signaling pathways including ERK 1/2 and NF-κB. Flecainide acetate also improved the survival rate in the rat sepsis model.
-
Conclusions
- Collectively, these findings indicate that flecainide acetate can improve survival in a rat sepsis model by attenuating LPS-induced neutrophil responses. We therefore suggest that flecainide acetate plays an important role in modulating inflammatory-immune responses.
-
Keywords: lipopolysaccharides; neutrophils; rats; sepsis; sodium channel blockers
Introduction
Despite the use of new interventions and modern antibiotics, sepsis is a common global health issue due to an aging population and mortality. The death rate in adult sepsis cases remains as high as 21%–35% [1-3]. Sepsis is defined as life-threatening organ failure caused by an uncontrolled host response to infection [4]. Neutrophils are the first cellular line of defense of the primary immune system to combat infection. They contribute 40%–60% of the leukocyte population in the blood and play a key role in the control of the immune system. At the infection site, neutrophils are recruited and create the first barrier of the host’s defense. Following stimulation, neutrophils synthesize and release important inflammatory mediators, including cytokines, chemokines, leukotrienes, and prostaglandins, which contribute to phagocytosis, bacterial killing, oxidative bursts, and degranulation [5,6]. In this way, neutrophils play a pivotal role in the occurrence of organ dysfunction related to serious infection. Nontoxic substances that control neutrophil-mediated inflammatory-immune responses could provide new methods for the treatment of sepsis.
Flecainide acetate, a potent sodium channel blocker, exerts electrophysiological effects to control supraventricular tachycardia and has been widely used in clinical settings. In addition to its electrophysiological effects, flecainide acetate affects autoimmune neutropenia [7] and the inhibition of human tumor necrosis factor-alpha (TNF-α) [8]. However, the ability of this sodium channel blocker to directly participate in neutrophil activation by lipopolysaccharide (LPS) has not been studied. In this study, we evaluated whether flecainide acetate decreases the activation of neutrophils induced by LPS and blocks the effects of intracellular signaling pathways, including mitogenactivated protein (MAPK) kinase, nuclear factor (NF)-κB, and proinflammatory cytokines (TNF-α, interleukin [IL]-6 and IL-8), on neutrophils. Additionally, septic rats treated with flecainide acetate were evaluated for mortality.
Materials and Methods
Animals
Male Sprague-Dawley rats, 8–12 weeks of age, were purchased from Damul Science (Daejeon, Korea). The rats were kept on a 12-hour light/dark cycle with free access to food and water. All experiments were conducted in accordance with Chonnam National University’s Institutional Animal Care and Use Committee.
Materials
Escherichia coli O11:B4 endotoxin (LPS) was purchased from Sigma-Aldrich (St. Louis, MO, USA). RPMI-1640 medium (containing 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) and L-glutamine), fetal bovine serum, and penicillin/streptomycin were obtained from Mediatech (Herndon, VA, USA). The bicinchoninic acid (BCA) protein assay reagent was obtained from Pierce (Rockford, IL, USA). Antibodies specific for p-p38, p-ERK, and p-JNK (c-Jun N-terminal kinase), as well as total p38, extracellular signal-regulated kinase (ERK), and JNK, were purchased from Cell Signaling Technology (Beverly, MA, USA).
Isolation of Neutrophils
For human neutrophil isolation, peripheral blood was obtained from healthy volunteers under a protocol approved by Institutional Review Board of Chonnam National University Hospital (IRB No. CNUH-2012-048). Neutrophils were isolated using plasma-Percoll gradients after dextran sedimentation of erythrocytes as described previously [9]. The neutrophil purity, as determined using Wright-stained cytospin preparations, was greater than 97%. The cell viability, as determined using trypan blue exclusion, was consistently greater than 98%.
Cytokine Enzyme-Linked Immunosorbent Assay
Immunoreactive TNF-α, IL-6 and IL-8 were quantified using commercially available enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions as described previously [10].
Western Blot Analysis
Western blots to detect the levels of phosphorylated and total p38, ERK, and JNK were performed essentially as previously described [10,11]. The neutrophils were lysed in ice-cold lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid [EDTA]-Na2, 1% Triton X-100, 1 mM ethylene glycol tetraacetic acid, 1 mM Na3 vanadate, 2.5 mM Na pyrophosphate, 1 mM β-glycerophosphate, 1 mM phenylmethanesulfonyl fluoride [PMSF], 1 μg/ml leupeptin) and sonicated for 30 seconds. The debris from the lysed cells was pelleted via centrifugation at 14,000 for 20 minutes. The supernatant was then removed and stored at –86°C. The protein concentration of each sample was assayed using the BCA protein assay kit (Pierce) standardized to bovine serum albumin (BSA) according to the manufacturer’s protocol. For the Western blots, 50 μg of protein was electrophoresed in each well of a 10% Tris-HCl sodium dodecyl sulfate-polyacrylamide gel. The proteins were then electrotransferred to a nitrocellulose membrane and blocked with 20 mM Tris-buffered saline containing 5% nonfat dry milk and 0.1% Tween 20. After blocking, the membrane was incubated overnight at 4°C with rabbit polyclonal primary antibodies specific to p-Akt, p-JNK, or p-p38 (1:1000 dilution) in 1% BSA, followed by an anti-rabbit or anti-rat immunoglobulin G horseradish peroxidase-coupled secondary antibody (1:2000 dilution) in 5% nonfat dry milk. After washing five times, the bands were detected using electrochemiluminescence Western blotting detection reagents (Amersham Pharmacia Biotech, Piscataway, NJ, USA). The membranes were then stripped using stripping buffer (63 mM Tris-HCl pH 6.8, 2% sodium dodecyl sulfate, 100 mM 2-ME; Bio-Rad, Hercules, CA, USA) and reprobed with antibodies specific to total ERK, JNK, and p38. Densitometry was performed using a chemiluminescence system and the included analysis software (Bio-Rad) to determine the ratio between the phosphorylated and total kinases.
Electrophoretic Mobility Shift Assay
Electrophoretic mobility shift assays were performed as previously described [10,12]. To obtain nuclear extracts from the neutrophils, the cells were resuspended in buffer. A containing 10 mM HEPES (pH 7.9), 1.5 mM MgCl2, 10 mM KCl, and 0.5 mM dithiothreitol, and the samples were incubated on ice for 20 minutes. After the cytoplasm was removed from the nuclei using 15 passages through a 25-gauge needle, the nuclei were collected using centrifugation at 600 ×g for 6 minutes at 4°C. The pellets were suspended in buffer C containing 20 mM HEPES, 1.5 mM MgCl2, 420 mM NaCl, 0.2 mM EDTA, 25% glycerol, and 0.5 mM PMSF. After a 30-minute incubation on ice, the suspension was centrifuged at 14,000 ×g for 20 minutes at 4°C, and the supernatant was collected. The protein concentration in the supernatant was determined using the BCA protein assay kit (Pierce). The nuclear extracts (5 μg) were incubated at room temperature for 20 minutes in 20 μl of reaction buffer (10 mM Tris-HCl pH 7.5, 1 mM MgCl2, 0.5 mM EDTA, 0.5 mM dithiothreitol, 50 mM NaCl, and 4% glycerol) with a 32P end-labeled, double-stranded oligonucleotide probe specific for the κB site (5’-AGTTGAGGGGACTTTCCCAGGC-3’; Geneka, Burlington, VT, USA) and 1 μg of poly(dI-dC)·poly(dI-dC). In some experiments, unlabeled NF-κB or cAMP responsive element binding protein oligonucleotides (Promega, Madison, WI, USA) were added to the samples at a 200-fold excess before the addition of the labeled probe and incubated for 15 minutes on ice. The complexes were resolved on 5% polyacrylamide gels in Tris-HCl (pH 8.0)-borate-EDTA buffer at 10 V/cm. The dried gels were exposed to Kodak Biomax MS film (Rochester, NY, USA) for 1–24 hours at –70°C.
In vivo Sepsis Model
Sepsis was induced via intraperitoneal (IP) administration of 25 mg/kg of O4:B111 E. coli endotoxin (LPS) into rats. Animals were randomly assigned to one of five groups: (1) subcutaneous (SC) infusion of saline and IP injection saline (S-S group, negative control, n = 10), (2) SC infusion of saline and IP injection of E. coli LPS (25 mg/kg) (S-E group, positive control, n = 10), (3) SC infusion of flecainide acetate (0.2 mg/kg/hr) and IP injection of saline (F-S group, n = 10), (4) SC infusion of flecainide acetate (0.1 mg/kg/hr) and IP injection of LPS (F0.1-E group, low-dose, n = 10), and (5) SC infusion of flecainide acetate (0.2 mg/kg/hr) and IP injection of LPS (F0.2-E group, high-dose, n = 10). The SC infusion of saline or flecainide acetate using a mini osmotic pump was initiated 3 hours before the IP injection of saline or LPS and continued until the end of the experiment. The mortality rates were calculated 24 hours after the IP injection of saline or LPS.
Statistical Analysis
The data are presented as the mean ± standard error for each experimental group. One-way analysis of variance and the Tukey-Kramer multiple comparisons test (for multiple groups) or the Student t-test (for comparisons between two groups) was used. Null hypotheses of no difference were rejected if the P-values were less than 0.05.
Results
The Effects of Flecainide Acetate on Cytokine Expression (TNF-α, IL-6 and IL-8) in LPS-Stimulated Neutrophils
To evaluate the effects of flecainide acetate on cytokine expression (TNF-α, IL-6 and IL-8) in LPS-stimulated neutrophils, neutrophils were cultured with or without LPS (100 ng/ml) and flecainide acetate (1, 10, or 100 μM) for 4 hours. The neutrophils stimulated with LPS had increased levels of inflammatory cytokines (TNF-α, IL-6 and IL-8) compared to those in the control or flecainide acetate groups. However, treatment with a combination of LPS and flecainide acetate significantly inhibited cytokine expression in neutrophils, specifically at a flecainide acetate concentration of 100 μM (Figure 1).
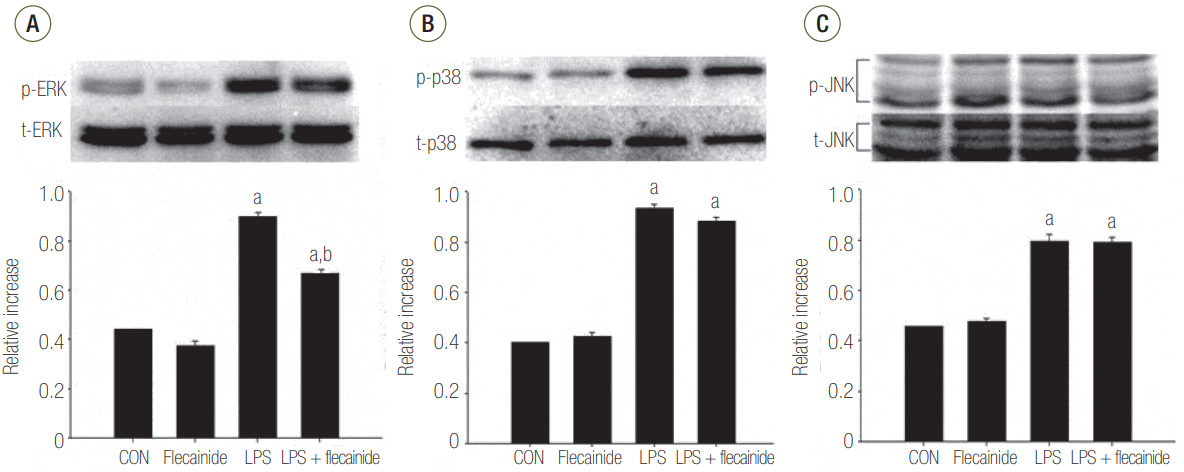
The Effects of Flecainide Acetate on MAPK Pathway Activation in LPS-Stimulated Neutrophils
Western blot analyses demonstrated notably higher protein expression of JNK, ERK 1/2, and p38 in the LPS group compared to the control or flecainide acetate groups. Treatment with a combination of LPS and flecainide acetate (100 μM) had no effect on the JNK or p38 pathways. However, the combination significantly reduced activation of the ERK 1/2 pathway compared with the LPS group (Figure 2).
The Effects of Flecainide Acetate on the Activation of NF-κB in LPS-Stimulated Neutrophils
To assess the effects of flecainide acetate on transcriptional regulation in LPS-stimulated neutrophils, we examined the nuclear levels of NF-κB in neutrophils cultured with LPS alone or with a combination of flecainide acetate and LPS. Incubation of neutrophils with LPS resulted in activation of NF-κB. The addition of flecainide acetate to the neutrophil cultures produced a decrease in the LPS-induced activation of NF-κB (Figure 3).
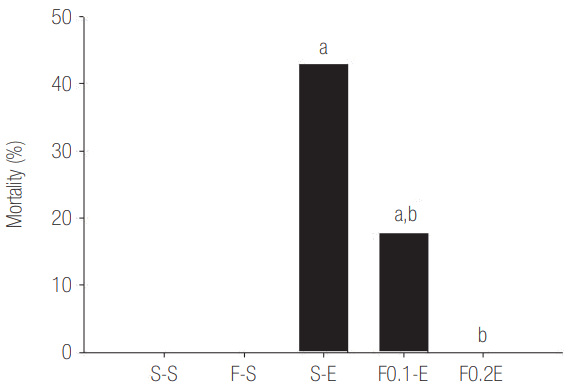
The Effects of Flecainide Acetate on Mortality in an LPS-Induced Rat Sepsis Model
To determine the effects of flecainide acetate in a rat sepsis model, rats were intraperitoneally injected with 25 mg/kg of LPS, and mortality was evaluated after 24 hours. Mortality was over 40% in the S-E (positive control) group, whereas mortality was markedly reduced to less than 20% in the F0.1-E (low-dose flecainide acetate) group. Interestingly, all rats in the F0.2-E (high-dose flecainide acetate) group survived (Figure 4).
Discussion
The main result of this study is that flecainide acetate has potential anti-inflammatory activities, including decreasing the activation of NF-κB and proinflammatory cytokines, in LPS-stimulated neutrophils in vitro. In additional in vivo studies, rats treated with flecainide acetate were protected from endotoxemia-induced sepsis as determined by mortality rates.
Binding sites for NF-κB are present in the promoter regions of the TNF-α, IL-6 and IL-8 genes, and NF-κB plays a pivotal role in the transcriptional control of each of these cytokines [10]. Additionally, p38 and ERK are involved in enhancing the activation of NF-κB and NF-κB-dependent genes in LPS-stimulated neutrophils [10,13].
Proinflammatory cytokines, such as TNF-α, IL-6 and IL-8, are major inflammatory mediators [14]. LPS-stimulated neutrophils up-regulate many kinases associated with the activation of proinflammatory cytokines. Among them, signaling pathways that bring about the activation of MAPKs (p38 and ERK 1/2) are especially important because the attenuation of MAPK-activation remarkably inhibits the activation of mediator cytokines that is induced by LPS [10,13,15]. Conversely, some studies also suggest that JNK, one of the members of the MAPK family, is activated in the proinflammatory events induced neutrophils [16,17]. Under these signaling pathways, neutrophils increase the release of oxygen radicals, proteases, leukotrienes, and proinflammatory cytokines (TNF-α, IL-1β and IL-8) [18-21]. Blocking the activation of these signaling pathways is a key strategy in the treatment of sepsis.
Flecainide acetate is an antiarrhythmic agent and functions as a sodium channel blocker. It also appears to modulate neutrophil action, suggesting that flecainide acetate may participate in controlling cellular recruitment during bacterial infection. This study extends the spectrum of activity of flecainide acetate to include anti-inflammatory effects on neutrophil activation.
In this study, the combination of LPS with flecainide acetate resulted in decreased activation of proinflammatory cytokines and ERK 1/2 compared with the group treated with LPS alone. In particular, flecainide acetate significantly reduced the activation of ERK 1/2, but not p38 or JNK, in LPS-stimulated neutrophils, suggesting that the effects on cytokine activation occur via an intracellular signaling pathway related to ERK 1/2 activation.
When primary neutrophils are exposed to inflammatory agents, such as bacterial LPS, it leads to the activation of transcriptional signaling pathways, including MAPK and NF-κB, resulting in the synthesis of cytokine mRNA and proteins [22,23]. Patients with sepsis have altered leucocytes and ERK and p38 kinase activation, which may be useful in detecting patients with sepsis and distinguishing them from those with systemic inflammatory response syndrome [24].
Our results showing that flecainide acetate inhibits neutrophil activation through an ERK 1/2-mediated pathway help explain why ERK 1/2 attenuation causes a decrease in the severity of conditions caused by neutrophil-associated acute sepsis such as acute respiratory distress syndrome.
The effects of flecainide acetate on important neutrophil functions, such as priming for superoxide production and chemotaxis, are almost reported. Therefore, this study’s major focus was to demonstrate whether flecainide acetate can attenuate the neutrophil activation induced by LPS under in vitro conditions.
The JNK and p38 MAP kinase pathways play an important role in inflammatory responses in LPS-stimulated neutrophils [13], but they are not involved in the alternation of neutrophil responses caused by flecainide acetate. Flecainide acetate did not affect neutrophil regulation through the p38 or JNK pathways. Coincubation of neutrophils with LPS and flecainide acetate did not result in decreased JNK and p38 activation. In contrast, LPS-induced phosphorylation of ERK 1/2 was remarkably attenuated when neutrophils were cultured with a combination with LPS and flecainide acetate. Inhibition of Na-H exchanger-1 by amiloride improved experimental colitis and was associated with the reversal of IL-1ss and ERK kinase [25]. Sikes et al. [26] also reported that the inhibition of Na/H exchange activity ameliorates sepsisinduced cardiac dysfunction. These findings suggest that the sodium channel blocking-effect of flecainide acetate plays a key role in reducing LPS-induced neutrophil activation.
In this study, we demonstrated the ability of flecainide acetate to attenuate neutrophil action in vitro and improve survival rates in an LPS-induced rat sepsis model. These effects suggest that flecainide acetate plays a major role in the regulation of neutrophil-mediated inflammatory responses.
NOTES
-
No potential conflict of interest relevant to this article was reported.
Acknowledgments
This study was supported by a grant (CRI12051-21) from the Chonnam National University Hospital Research Institute of Clinical Medicine.
Figure 1.The effects of flecainide acetate on proinflammatory cytokine (A: IL-6, B: IL-8, C: TNF-α) expression in LPS-stimulated neutrophils. Neutrophils (5 × 106/ml) from human blood were incubated for 4 hours with or without flecainide acetate (1, 10, or 100 μM) and with or without LPS (100 ng/ml) with the control group (CON) receiving no treatment. The protein levels were obtained using enzyme-linked immunosorbent assays. A representative experiment, in which each condition was examined in triplicate, is shown. Two additional experiments provided similar results. The data are presented as mean ± standard error for each experimental group. IL: interleukin; LPS: lipopolysaccharide; TNF-α: tumor necrosis factor-alpha. aP < 0.05, vs. CON; bP < 0.05, vs. LPS.

Figure 2.The effects of flecainide acetate on mitogen-activated protein (A: ERK, B: p38, C: JNK) pathway activation in LPS-stimulated neutrophils. Neutrophils (5 × 106/ml) from human blood were incubated for 30 minutes with or without flecainide acetate (100 μM) and with or without LPS (100 ng/ml) with the control group (CON) receiving no treatment. The histogram shows mean ± standard error from three experiments. ERK: extracellular signal-regulated kinase; LPS: lipopolysaccharide; p38: p38 mitogen-activated protein kinase; JNK: Jun N-terminal kinase. aP < 0.05, vs. CON; bP < 0.05, vs. LPS.
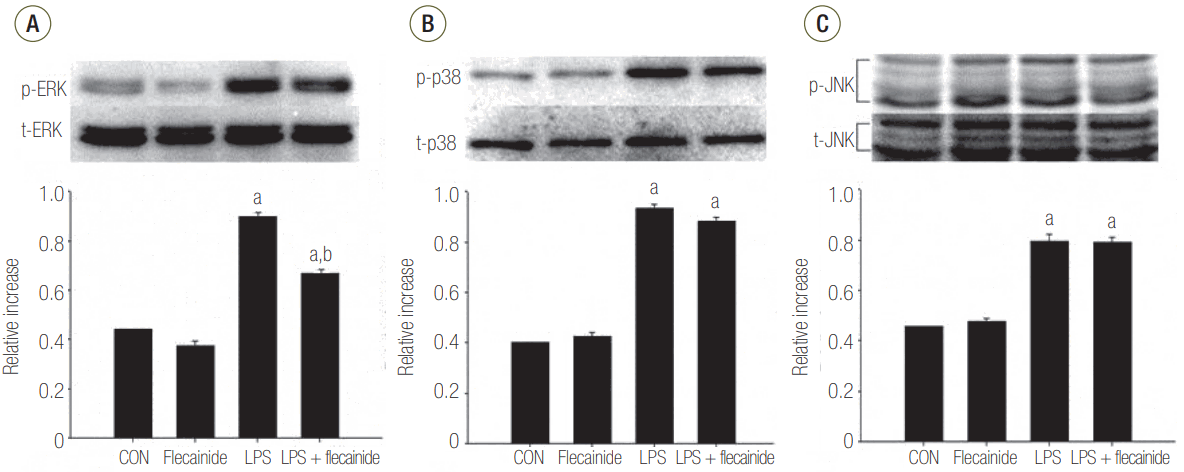
Figure 3.The effects of flecainide acetate on the nuclear translocation of nuclear factor (NF)-κB in lipopolysaccharide (LPS)-stimulated neutrophils. Neutrophils (5 × 106/ml) from human blood incubated for 1 hour with 100 ng/ml of LPS (LPS) showed more nuclear translocation of NF-κB than neutrophils incubated for 1 hour without LPS (Control) or with 100 μM flecainide acetate (Flecainide). Coincubation of neutrophils with 100 μM flecainide acetate and 100 ng/ml LPS (LPS + flecainide) resulted in less nuclear translocation of NF-κB compared to LPS alone. Three replicate experiments provided similar results.

Figure 4.The effects of flecainide acetate on mortality in a lipopolysaccharide-induced rat sepsis models. The mortality rates were calculated 24 hours after the intraperitoneal (IP) injection of saline or lipopolysaccharide. Mortality was over 40% in the S-E (positive control) group, whereas mortality was markedly reduced to less than 20% in the F0.1-E (low-dose flecainide acetate) group. All rats in the F0.2-E (high-dose flecainide acetate) group survived. Each value represents mean ± standard error from 10 rats. S-S: subcutaneous (SC) infusion of saline and IP injection saline (n = 10); F-S: SC infusion of flecainide acetate (0.2 mg/kg/hr) and IP injection of saline (n = 10); S-E: SC infusion of saline and IP injection of Escherichia coli endotoxin (25 mg/kg, n = 10); F0.1-E: SC infusion of flecainide acetate (0.1 mg/kg/hr) and IP injection of endotoxin (n = 10); F0.2-E: SC infusion of flecainide acetate (0.2 mg/kg/hr) and IP injection of endotoxin (n = 10). aP < 0.05, vs. the S-S negative-control group; bP < 0.05, vs. the S-E positive-control group.

References
- 1. Ferrer R, Martin-Loeches I, Phillips G, Osborn TM, Townsend S, Dellinger RP, et al. Empiric antibiotic treatment reduces mortality in severe sepsis and septic shock from the first hour: results from a guideline-based performance improvement program. Crit Care Med 2014;42:1749-55.ArticlePubMed
- 2. Rhee C, Dantes R, Epstein L, Murphy DJ, Seymour CW, Iwashyna TJ, et al. Incidence and trends of sepsis in US hospitals using clinical vs claims data, 2009-2014. JAMA 2017;318:1241-9.ArticlePubMedPMC
- 3. van Zanten AR, Brinkman S, Arbous MS, Abu-Hanna A, Levy MM, de Keizer NF, et al. Guideline bundles adherence and mortality in severe sepsis and septic shock. Crit Care Med 2014;42:1890-8.ArticlePubMed
- 4. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 2016;315:801-10.ArticlePubMedPMC
- 5. Doerschuk CM, Tasaka S, Wang Q. CD11/CD18-dependent and -independent neutrophil emigration in the lungs: how do neutrophils know which route to take? Am J Respir Cell Mol Biol 2000;23:133-6.ArticlePubMed
- 6. Windsor AC, Mullen PG, Fowler AA, Sugerman HJ. Role of the neutrophil in adult respiratory distress syndrome. Br J Surg 1993;80:10-7.ArticlePubMed
- 7. Samlowski WE, Frame RN, Logue GL. Flecanide-induced immune neutropenia: documentation of a hapten-mediated mechanism of cell destruction. Arch Intern Med 1987;147:383-4.ArticlePubMed
- 8. Nojima S, Kobuchi H, Saibara T, Yasuda T, Utsumi K. Selective inhibition of human TNF-alpha action by flecainide acetate, an antiarrhythmic drug. Physiol Chem Phys Med NMR 1995;27:77-89.PubMed
- 9. Kwak SH, Mitra S, Bdeir K, Strassheim D, Park JS, Kim JY, et al. The kringle domain of urokinase-type plasminogen activator potentiates LPS-induced neutrophil activation through interaction with αVβ3 integrins. J Leukoc Biol 2005;78:937-45.ArticlePubMed
- 10. Yum HK, Arcaroli J, Kupfner J, Shenkar R, Penninger JM, Sasaki T, et al. Involvement of phosphoinositide 3-kinases in neutrophil activation and the development of acute lung injury. J Immunol 2001;167:6601-8.ArticlePubMed
- 11. Arcaroli J, Yang KY, Yum HK, Kupfner J, Pitts TM, Park JS, et al. Effects of catecholamines on kinase activation in lung neutrophils after hemorrhage or endotoxemia. J Leukoc Biol 2002;72:571-9.ArticlePubMedPDF
- 12. Harris VK, Danda D, Murali NS, Das PK, Abraham M, Cherian AM, et al. Unusual association of Kikuchi’s disease and dengue virus infection evolving into systemic lupus erythematosus. J Indian Med Assoc 2000;98:391-3.PubMed
- 13. Nick JA, Avdi NJ, Young SK, Lehman LA, McDonald PP, Frasch SC, et al. Selective activation and functional significance of p38α mitogen-activated protein kinase in lipopolysaccharide-stimulated neutrophils. J Clin Invest 1999;103:851-8.ArticlePubMedPMC
- 14. Baigrie RJ, Lamont PM, Kwiatkowski D, Dallman MJ, Morris PJ. Systemic cytokine response after major surgery. Br J Surg 1992;79:757-60.ArticlePubMed
- 15. Selvatici R, Falzarano S, Mollica A, Spisani S. Signal transduction pathways triggered by selective formylpeptide analogues in human neutrophils. Eur J Pharmacol 2006;534:1-11.ArticlePubMed
- 16. Avdi NJ, Nick JA, Whitlock BB, Billstrom MA, Henson PM, Johnson GL, et al. Tumor necrosis factor-alpha activation of the c-Jun N-terminal kinase pathway in human neutrophils: integrin involvement in a pathway leading from cytoplasmic tyrosine kinases apoptosis. J Biol Chem 2001;276:2189-99.ArticlePubMed
- 17. Rane MJ, Carrithers SL, Arthur JM, Klein JB, McLeish KR. Formyl peptide receptors are coupled to multiple mitogen-activated protein kinase cascades by distinct signal transduction pathways: role in activation of reduced nicotinamide adenine dinucleotide oxidase. J Immunol 1997;159:5070-8.ArticlePubMedPDF
- 18. Goebel LJ, Crespo RD, Abraham RT, Masho SW, Glover ED. Correlates of youth smokeless tobacco use. Nicotine Tob Res 2000;2:319-25.ArticlePubMedPDF
- 19. Esmon CT, Fukudome K, Mather T, Bode W, Regan LM, Stearns-Kurosawa DJ, et al. Inflammation, sepsis, and coagulation. Haematologica 1999;84:254-9.PubMed
- 20. Hardaway RM. A review of septic shock. Am Surg 2000;66:22-9.ArticlePubMedPDF
- 21. Karima R, Matsumoto S, Higashi H, Matsushima K. The molecular pathogenesis of endotoxic shock and organ failure. Mol Med Today 1999;5:123-32.ArticlePubMed
- 22. Lien E, Ingalls RR. Toll-like receptors. Crit Care Med 2002;30(1 Supp):S1-11.Article
- 23. Pace JL, Russell SW. Activation of mouse macrophages for tumor cell killing. I. Quantitative analysis of interactions between lymphokine and lipopolysaccharide. J Immunol 1981;126:1863-7.ArticlePubMedPDF
- 24. West MA, Koons A, Crandall M, Skinner R, Worley M, Shapiro MB. Whole blood leukocyte mitogen activated protein kinases activation differentiates intensive care unit patients with systemic inflammatory response syndrome and sepsis. J Trauma 2007;62:805-11.ArticlePubMed
- 25. Khan I, Oriowo MA, Anim JT. Amelioration of experimental colitis by Na-H exchanger-1 inhibitor amiloride is associated with reversal of IL-1ss and ERK mitogen-activated protein kinase. Scand J Gastroenterol 2005;40:578-85.ArticlePubMed
- 26. Sikes PJ, Zhao P, Maass DL, White J, Horton JW. Sodium/hydrogen exchange activity in sepsis and in sepsis complicated by previous injury: 31P and 23Na NMR study. Crit Care Med 2005;33:605-15.ArticlePubMed
Citations
Citations to this article as recorded by

- Persistence is key: unresolved immune dysfunction is lethal in both COVID-19 and non-COVID-19 sepsis
Andy Y. An, Arjun Baghela, Peter Zhang, Reza Falsafi, Amy H. Lee, Uriel Trahtemberg, Andrew J. Baker, Claudia C. dos Santos, Robert E. W. Hancock
Frontiers in Immunology.2023;[Epub] CrossRef




 PubReader
PubReader ePub Link
ePub Link Cite
Cite